SPP 2363 on “Utilization and Development of Machine Learning for Molecular Applications – Molecular Machine Learning”
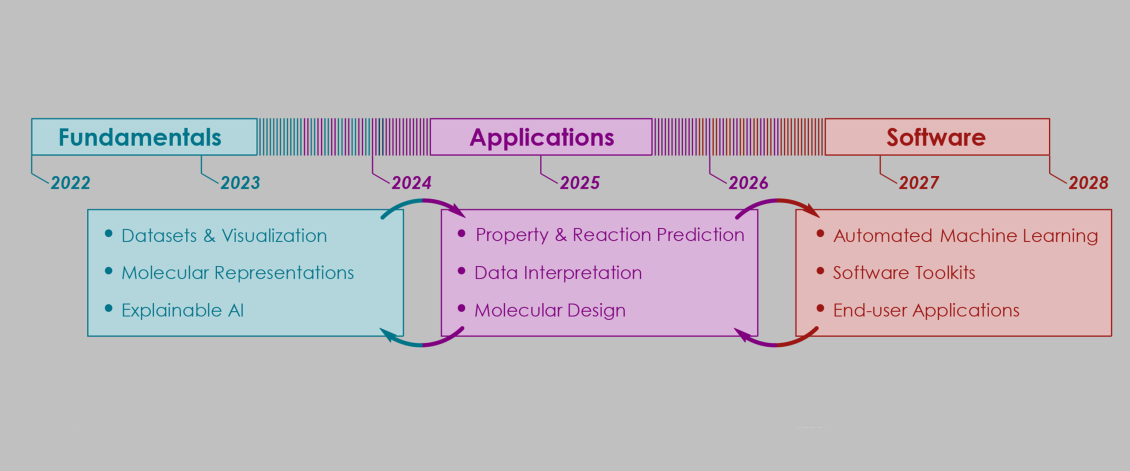
From Fundamentals to Application and Beyond
“Today the computer is just as important as tool for chemists as the test tube. Simulations are so realistic that they predict the outcome of traditional experiments.”
Staying abroad with the SPP2363 – Computational Drug Design at the National Autonomous University of Mexico
A short report by Samuel Homberg, Koch Group, University of Münster
Thanks to the SPP2363 scholarship I was able to spend some of the most exciting time of my PhD at Professor Medina-Franco's group DIFAQUIM at the National Autonomous University of Mexico. I got to research different methodologies in cheminformatics and focused on combining (machine-learning based) molecular representations, fitting well into the scope of the SPP. It was great to get an insight into how the group explores chemical space using a vide variety of tools, sometimes with downright artistic visualizations. During this time I was also able to write my first Python package and extend my coding skills.
Finally, I was also able to spend two weeks at UNAMs offshoot in Mérida under the supervision of Norberto Sánchez-Cruz, working on a novel neural fingerprint representation (different from my own project on neural fingerprints), giving me new input and ideas.
I am extremely grateful to the SPP2363 that I not only got to learn a lot and build my professional network but I also got to immerse myself in a different culture and I made a bunch of new friends.
Selection of projects for the second funding period of SPP 2363
A review colloquium to select projects for the next funding phase will take place in Münster early next year.
Venue: Centre for Soft Nanoscience (SoN), Busso-Peus-Straße 10, 48149 Münster, Germany.
Start: 29.01.2025 at 10am,
End: 29.01.2025 at 16:00.
In order to give the jury a comprehensive overview, it is planned that all applicants will present their projects in short presentations of max. 3 minutes at the beginning of the colloquium. To ensure a smooth process, please send your presentation (max. 3 slides) as a PDF file to the coordinator (send to: glorius@uni-muenster.de) by 22 January. Following the short presentations, there will be the opportunity to present the planned projects of the study group in detail and to answer questions in a poster session.
SPP2363 PI Matthias Rarey, University of Hamburg, receives the 2025 Herman Skolnik Award of the ACS, CINF Division
The American Chemical Society Division of Chemical Information announced that Prof. Matthias Rarey has been selected to receive the 2025 Herman Skolnik Award for his contributions to the development of foundational algorithms in Cheminformatics, education & training in the field of Cheminformatics and his activities bridging academia and industry.
Prof. Rarey’s research concentrates primarily on methods for ligand-based and structure-based molecular design. His early work focused on the development of novel techniques for flexible ligand-protein docking and his postdoctoral research resulted in the Feature Tree concept, an approach to the representation and searching of small molecules that has found particular application for exploring combinatorial chemistry and fragment spaces. More recent studies have led to novel approaches for the drawing and visualization of molecular structures, chemical patterns, and biological macromolecules, for de novo design, for the analysis of protein binding sites and torsion-angle distributions, and for 3D shape similarity. Examples of systems derived from his research include the FlexX protein-ligand docking program, PoseView for generating 2D diagrams of complexes with known 3D structures, FTrees for similarity searching in chemical spaces, HYDE for scoring hydrogen bond and dehydration energies in protein-ligand complexes, and ReCore for scaffold-hopping in lead-discovery programs. Recently, SpaceLight and SpaceMACS were added enabling topological similarity searching in combinatorial make-on-demand catalogues like Enamine REAL for the first time. Within SPP2363, Matthias Rarey and Frank Glorius work together on the synthetically feasible extension of chemical fragment spaces with machine learning (Project SAFE).
Staying abroad with the SPP2363 – High-Throughput Screening and Chemoinformatics at the University of Michigan
A short report by Felix Katzenburg, Glorius Group, University of Münster
With the support of an SPP scholarship, I was able to join the Cernak group at the University of Michigan for a 6-month research stay. It was an amazing experience to learn new techniques for high-throughput experimentation, library synthesis and direct-to-biology approaches. It was great to see how the group collectively used expertise in synthesis, chemoinformatics, software development, medicinal chemistry, and biology to identify and synthesize molecular libraries for testing in biological assays. Personally, I had the chance to advance the concept of late-stage saturation, which was introduced in a collaborative project with our group in Germany to facilitate library synthesis.
In addition, I had the chance to work on chemoinformatic approaches in reaction enumeration to identify and prioritize valuable and synthetically relevant reactions that are yet to be developed. I am very thankful for the warm welcome and support I received from the entire Cernak group during my stay. Besides the fantastic research opportunities, I am also really grateful for the chance to explore Michigan and the US and build many friendships in the process. I would like to thank Prof. Tim Cernak and the SPP2363 for this great opportunity.
This programme aims at connecting communities from the fields of machine learning and data science with scientists working in the areas of molecular chemistry and pharmacology. Machine learning for molecular applications and questions (Molecular Machine Learning, MML) has emerged as an area of interest with a high potential to change current workflows in all fields of chemistry as well as pharmacology. As such, it poses several outstanding challenges. This Priority Programme aims at tackling these challenges in a holistic fashion, covering a spectrum of topics ranging from data generation and the application of new algorithms to explainable artificial intelligence (ExAI). In general, all projects are required to contribute to the whole MML community by developing reusable tools, methodologies, datasets or broadly utilisable applications. Each proposal must be positioned at the interface of chemistry/pharmacology and machine learning in at least one of the following five areas:
design and evaluation of molecular representations for machine learning;
machine learning as a tool for theoretical and organic chemistry;
machine learning for medicinal chemistry and drug design;
overcoming data limitations by data generation, evaluation and data-free approaches;
development of machine learning tools for molecular applications including ExAI, data augmentation strategies and software suites.
Thanks to the SPP scholarship, I had the amazing opportunity to spend three months on a research stay abroad at the University of Michigan in the United States. It was an absolute pleasure to work alongside Prof. Timothy Cernak and his group. The group is made up of talent from a variety of disciplines, including chemists, pharmacists and software engineers. Their focus on high-throughput synthesis was particularly relevant to my research on reaction networks for synthesis. We also started a project in collaboration with Prof. Paul Zimmerman's group to investigate the use of large language models (LLMs) in predicting activation energies to aid reaction discovery. We used techniques such as one-shot and few-shot learning, as well as fine-tuning of different LLMs using previously published work from the Zimmerman group on reaction discovery computations on B, C, N, O ring formation using NH3BH3 and CO2 as data.
While research was a priority, the university offered a lot of different recreational activities that I was able to enjoy with my newly found friends. I am sure that this collaboration will continue in the future. Thanks again to the SPP 2363 to make all of this a reality, it was an incredible experience!
Staying abroad with the SPP2363 - Machine Learning at the University of Edinburgh, UK
A short report by Felix Moorhoff, Davari Group, Leibniz Institute of Plant Biochemistry Halle/Saale
I am very grateful to have been granted the SPP2363 scholarship for a research stay in the group of Dr. Antonia Mey at the University of Edinburgh, UK. During the 6 weeks I had the fantastic opportunity to learn and adapt Machine Learning concepts from the field of Drug Discovery onto enzymatic catalysis prediction. The Mey group is well known for its explorations of Artificial Intelligence techniques on biomolecular modelling. I was highly impressed by the group’s expertise and particularly enjoyed a close collaboration with Rohan Gorantla who is a final year PhD student in the group. During my visit we explored new concepts that will help with enzymatic catalysis ML tasks. Together, we developed and applied concepts of Positive Unlabeled Learning on the prediction of enzyme reactions. This is now being benchmarked on existing tools and we are hoping for a joint publication. In the next step we want to explore Active Learning strategies for enzyme discovery campaigns to evaluate proteins fitness and usability for small molecule enzymatic catalysis. We are all looking forward to further collaboration and are very grateful for the opportunity provided by SPP2363.
International Leopoldina Symposium in Halle (Saale) 2024
The international Symposium on Molecular Machine Learning took place from 04.03 – 06.03 2024 in the German National Academy of Sciences Leopoldina. More than 170 participants from academia, as well as industry, joined the event and learned about the latest advancements in molecular machine learning. The symposium brought together world-leading scientists from diverse fields of molecular machine learning. Spanning three inspiring days, the symposium featured a rich program comprising 15 fascinating lectures that explored a wide spectrum of topics, ranging from innovative strategies to accelerate drug discovery to the cutting-edge developments in self-driving laboratories and advancements in efficient quantum mechanics (QM) calculations. These lectures not only provided valuable insights into the current state of the field but also sparked stimulating discussions and debates among the participants.
In addition to the lectures, the symposium featured two interactive poster sessions, offering participants an opportunity to presenting their own research activities and engage in thought-provoking discussions with peers. The Leopoldina, with its grandeur and historical significance, provided an amazing setting for gaining new knowledge, connecting with many researchers, and exchanging and discussing new ideas. We thank all participants and helpers who contributed to making this symposium a great success.
6th International Mini-Symposium Molecular Machine Learning, January 18th 2024
The mini-symposium series on “Molecular Machine Learning” went to the 6th round. More than 200 participants joined the symposium and made it yet again a very special event. Francesca Grisoni (Eindhoven University of Technology), Jens Meiler (University of Leipzig & Vanderbilt University), Franziska Schoenebeck (RWTH Aachen) and Fred Manby (Iambic Therapeutics) shared their vision for accelerating drug discovery and homogeneous catalysis and presented their strategies for dealing with the present data shortage in chemistry.
The SPP2363 scholarship for research abroad allowed me to stay almost three months at the Massachusetts Institute of Technology in Cambridge MA, USA. This opportunity helped me to grow not only scientifically but also personally. I spend my time at the MIT in the group of Prof. Rafael Gómez-Bombarelli (short: Rafa) in the department of material science and engineering. Rafa is an established professor for computer enhanced molecular discovery and design as well as molecular machine learning. Part of his research focus lays on machine learning potentials, generative models and fundamental science related to molecular machine learning. All these fields are perfectly in line with my own PhD project in the SPP2363.
During my stay I worked on ML potentials to speed up transition state search algorithms. The final goal is to predict highly accurate transition state barriers in a fraction of the ab initio calculation time. My direct supervisor during my stay was Dr. Johannes Dietschreit, a postdoc in Rafa’s group. In plenty of discussions Johannes explained to me the different approaches to generate a dataset, training of the potential and active learning cycles to achieve my goal of building a functioning ML potential. In the process of trying out the different approaches my overall understanding of this area of machine learning, which was new to me before my research stay, drastically increased. In addition to improvements in my own PhD project, I was able to learn a lot about the broad range of research projects in Rafa’s group. Either by personal scientific discussions or based on group meeting talks on a weekly basis, I learned a lot about general concepts of molecular machine learning and challenges that can occur while studying them.
The scientific aspects made my research stay to a memorable experience. On top of that, I was able to grow personally based on rich cultural exchange and the variety of activities that Cambridge and Boston had to offer.
Molecules Meet Materials
A short report by Felix Katzenburg, Group of Prof. Frank Glorius, University of Münster
Invited by SPP2363 member and Virtual Materials Design (VirtMat) coordinator Prof. Wolfgang Wenzel, I had the great opportunity to present my research on "Data-driven Organic Chemistry" in the VirtMat seminar at the Karlsruhe Institute of Technology. Thanks to the hospitality of Niklas Kappel and the entire Wenzel group my visit was a fun opportunity to connect within the SPP. Great discussions with the groups of Prof. Levkin and Prof. Bräse also gave me a chance to gain some insights into innovative research in miniaturization, data management, and automation beyond our priority program.
Many thanks to Prof. Wenzel, Niklas Kappel and the Levkin and Bräse groups for a great experience.
International Leopoldina Symposium in Halle (Saale) 2024
From March 04th till March 06th we’ll host an International Leopoldina Symposium in Halle (Saale). The symposium will take place in the German National Academy of Sciences Leopoldina. During three inspiring days, international leading scientists from diverse fields of molecular machine learning will share their research. A particular focus of the symposium is on topics such as the interpretability of model predictions, the development of automated laboratories and the use of ML for the accelerated development of functional materials and drugs. Besides insightful lectures, there will be poster sessions where participants are invited to present their research, engage in discussions and exchange ideas. Participation is free of charge, but the number of participants is limited. However, registration is required, please find more informations here.
The sixth edition of the international symposium series on "Molecular Machine Learning" organized by Prof. Frank Glorius will take place on January 18th, 2024 from 3.00 pm. Four inspiring speakers will share their research and perspective on various fields of molecular machine learning, including ML for drug discovery using small data, ultra-large library screening and ML assisted catalyst discovery. The virtual conference will be hosted on Zoom and participation is free of charge. However, registration is required, please find more informations here.
Staying abroad with the SPP2363 – Machine Learning at Vanderbilt University, USA
A short report by Fabian Liessmann, Group of Prof. Jens Meiler, University of Leipzig
Thanks to the scholarship for PhD students to stay abroad of the SPP2363, I had the opportunity to expand my research projects for several weeks at the Vanderbilt University, Nashville, Tennessee, USA. During that time, I worked together with several members of our partner lab, and especially, with the research assistant professor Benjamin (Ben) Brown, who specialized in the development and application of computer-aided drug design methods. Ben Brown is one of the main developers and contributors of the BioChemical Library, in short BCL, which employs machine learning algorithms for more than 15 years. Furthermore, he is an expert for optimizing drug candidates with both traditional and deep learning methods. In many fruitful discussions, we evaluated the current paradigm shift in the field and discussed the future of machine learning, especially for small molecules. Furthermore, during a special session about “Recent Developments and Technologies in ML”, I had the opportunity to listen to presentations from various researchers and directly discuss it with the lab members. A highlight was definitely the talk “Towards Data-Centric Graph Learning for Real-World Applications” from Assistant Professor Tyler Derr from Vanderbilt, where he presented his current research projects and machine-learning approaches.
This internship in Nashville gave me valuable insights outside of my day-to-day research environment and work. Of course, the personal and direct communication facilitates the exchange of research ideas, especially during hiking or group events in Nashville. The international lab, its environment and the welcoming/open nature of America captured and embraced me and round off an exciting time. I am really grateful to the SPP2363 for this unique and rewarding opportunity.
This year’s annual meeting of the SPP 2363 took place at the Friedrich-Schiller University of Jena from the 12th to the 14th of September 2023. Several lectures and a poster session updated everyone on the current status of the research projects as well as providing an interactive setting for exchanging and discussing new ideas. In addition to the research project updates from all members of the SPP 2363, there were also workshops on gender & diversity in science as well as a hands-on introduction to robot-based chemistry. Furthermore, two speakers from industry gave fascinating insights into automation and data management. Overall, the meeting was a great success by strengthening connections, exchanging ideas and forming new synergies.
The PhD students of the SPP 2363 participated in a workshop on scientific presentations on the 22nd and 23rd of August 2023 in Münster. Led by the experienced trainer Dr. Alexander Britz the students enhanced their skills for effective scientific communication with a special focus on oral and poster presentations. In interactive and fun exercises the participants learned techniques to present their scientific results in a structured and appealing way.
Fabian Jirasek is granted the new Emmy Noether Junior Research Group "Hybrid Thermodynamic Models" by the German Research Foundation (DFG), which will be established at RPTU Kaiserslautern. The group will develop novel thermodynamic models to predict the fluid properties of mixtures. The knowledge of these properties is essential for the chemical industries, e.g., for process optimization and establishing new production routes that rely on renewable instead of fossil raw materials.
Mixtures are omnipresent when producing chemicals and medicines, but also in developing new batteries, e.g., for e-cars. "Understanding the properties of these mixtures is of central importance for basically all processes in chemical engineering, from the reaction to the purification of the target products," says Fabian Jirasek, who is a Junior Professor for Machine Learning in Process Engineering at RPTU Kaiserslautern. "They are the basis for design and optimization of efficient processes."
Investigating all conceivable combinations of substances and the influence of parameters, such as temperature and pressure, in laboratory experiments is not feasible due to the abundance of possibilities. "Therefore, we rely on our models to also predict the fluid properties of non-measured substances and mixtures as well as at non-measured states," explains Jirasek. In thermodynamics, such models have been established for decades. But thanks to machine learning (ML), a subfield of artificial intelligence, research today has entirely new possibilities. "ML techniques will revolutionize thermodynamic modeling," Jirasek is certain. In the new project, Jirasek and his team will develop hybrid models combining machine learning and physical modeling. "We assume that machine-learning methods will require significantly less data by exploiting the available physical knowledge," he continues. "This will also create trust and acceptance for the novel models if they or their predictions satisfy known physical laws."
The DFG's Emmy Noether Programme is aimed at outstandingly qualified young researchers. The goal is that they can thereby qualify for a professorship by independently leading a junior research group.
Fabian Jirasek studied bioengineering at the Karlsruhe Institute of Technology and earned his doctorate in thermodynamics at Kaiserslautern. He then researched at the University of California at Irvine before moving to TU Munich. Since the fall of 2020, he has held the Junior Professorship for Machine Learning in Process Engineering at RPTU Kaiserslautern, funded by the Carl Zeiss Foundation.
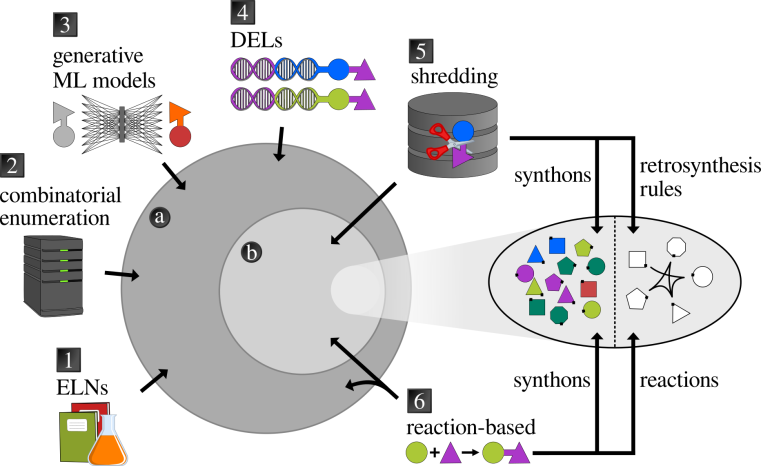
The use of chemical spaces for drug discovery has become a major focus in the scientific community, highlighting their growing importance. In a recent review paper, co-authored by Malte Korn from the SPP 2363 and published in Current Opinion in Structural Biology, a detailed examination of the current state of chemical spaces is given. The article covers the fundamentals of large chemical spaces and their importance for drug discovery making it an excellent introductory source for non-experts.
So, if you are new to this field start your journey through chemical spaces with this open-access review today!
The latest semiempirical tight-binding method for electronic structure calculations by the Grimme group (S. Grimme, M. Müller, and A. Hansen) has made it to the cover of The Journal of Chemical Physics! The method, named PTB, is excellently suited for the generation of descriptors in the context of modern machine-learning approaches based on quantum-chemical features. As PTB is available for all elements up to Radon (except for lanthanides) and is a non-self-consistent two-step method, it provides consistent and robust results throughout large parts of the chemical space. The method will be publicly available in the open-source xtb program package - give it a try!
5th International Mini-Symposium Molecular Machine Learning, January 19th 2023
Yesterday, the fifth edition of the international symposium series on "Molecular Machine Learning" took place virtually. Over 200 participants joined the symposium and made it yet again a very special event. The invited speakers Núria López (ICIQ), Kim Jelfs (Imperial College London), Tim Cernak (University of Michigan) and Sarah Reisman (Caltech) gave fascinating insights into their research and highlighted how data-driven approaches can be utilized for computer-aided synthesis planning, molecular design, automation and drug discovery.
The fifth edition of the international symposium series on "Molecular Machine Learning" organized by Prof. Frank Glorius will take place on January 19th, 2023 from 3:00 pm. The virtual conference series brings together leading scientists from fields including computer-aided synthesis planning, data-driven molecular design, automation and AI-enabled drug discovery. Speakers at the symposium will be: Núria López (ICIQ), Kim Jelfs (Imperial College London), Tim Cernak (University of Michigan) and Sarah Reisman (Caltech).
SPP 2363 Kick-off Symposium
The SPP 2363 Kick-off Symposium took place at the German National Academy of Sciences Leopoldina in Halle from October 17th to October 21st. With PhD talks and posters on the various applications of molecular machine learning anchored in the priority program and the opportunity to build an even stronger the community, the event was a great success. Talks from industry and researcher provided a great setting for exchanging ideas and setting goals for the future development of the program. We were also very pleased that Tiago Rodrigues (University of Lisbon) accepted our invitation and joined the symposium as the first SPP 2363 Mercator Fellow. The event was supported by the German National Academy of Sciences Leopoldina and the DFG.
SPP 2363 officially started
The DFG has selected suitable projects and the official funding letters were received. Let' get started, everyone!
Interested students, kindly contact the respective principal investigators.
In C&EN's June cover story, Matthias Rarey highlights how computational tools help to navigate chemical spaces and virtual libraries in the search for new drugs.
Connecting chemical building blocks allows drug hunters to explore a much bigger chemical space than before. The challenge is to narrow this field of compounds to something manageable. To do that, chemists are turning to new computational tools to navigate this increasingly huge chemical universe, and they are combining technologies. Experts say these new approaches should speed up the identification process, and industry is investing time and money to optimize the hunt.
Frank Glorius and Philipp Pflüger talk about the new field of research “Molecular Machine Learning".
“Molecular Machine Learning” (MML) is a new branch of research with the potential to change chemical research. Prof. Frank Glorius, coordinator of the new Priority Programme “Molecular Machine Learning” (SPP 2363), funded by the German Research Foundation (DFG), and Philipp Pflüger, who is working on his PhD in Chemistry and helped to develop the programme, explain in this interview with Christina Hoppenbrock what MML means, what opportunities and challenges this new field of research presents, and what working in chemistry will be like in tomorrow’s world.