
Laser desorption of small or diatomic gas molecules from a C60/Cu(111) surface
A C60-molecule (also called “Buckminster-fullerene”)[1] consists of sixty carbon atom ordered in a structure of twelve pentagons and 20 hexagons, similar to the structure of an usual football. Its existence was predicted in 1970 by E. Osawa[2], but not until 1985 the production of fullerene powder was enabled by H.W. Kroto et al.[1]. Until today the chemical, mechanical and eletronic properties are observed, and C60 was identified as semiconducting material ordered in a layered array. Because of this C60 is an advantageous sample for investigating interactions of gases with solids and catalysis processes on a C60 surfaces.
Electronic states
Copper is a metal, but the (111)-surface has a directional band gap, reflecting electrons in normal incidence in the energy range: -0.8...4.0 eV.
Low electronic states in C60 get about 0.4 eV band dispersion when condensed. This leads to no overlap, and is ignored in the following discussion:
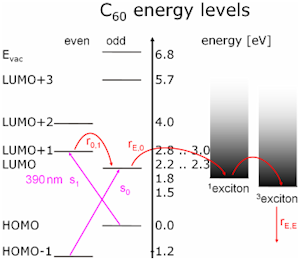
The Highest Occupied Molecular Orbitals and Lowest Unoccupied Molecular Orbitals are electronic states in single-particle theory grouped by symmetry. For example for the electrons of the He atom it is a too crude approximation as spin-spin-coupling must be observed which belongs to many-particle theory. Let’s assume that someone was able to calculate the ground state of bulk semiconductors with many-particle theory. Then single-particle theory uses annihilation and creation operators to annihilate an electron (create a hole) and create an excited electron. Excitons are two particle states of one hole and one electron and represent a step towards many-particle states, adding minor corrections the density of states. The lowest triplet exciton is part of the decay channel of most excitons, has a µs lifetime and so is highly occupied. Therefore and because it lies in the band gap it is visible in many experiments. In a small band gap semiconductor excitons are hydrogen like systems and can be reformulated as a single particle problem without any approximations (Wannier exciton) in contrast to the Frenkel excitons in C60 .
Experimental setup
The setup of the desorption experiment consists of a desorption laser system, an ionization/detection laser system and the vacuum chamber, which includes several tools for priming and analyzing the sample.
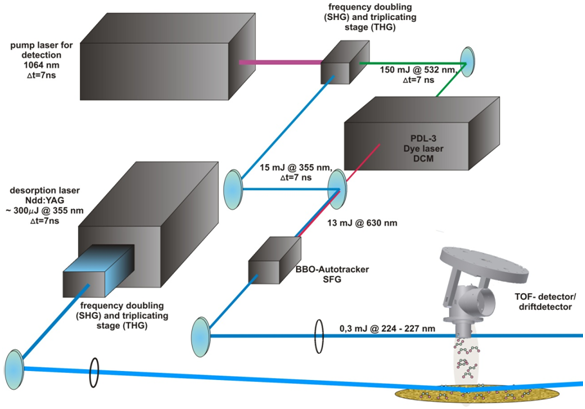
The desorption laser system (fig. 2) consists of a Nd:YAG Laser, which emits infrared pulsed radiation (10 Hz @ 1064 nm) and is directly frequency doubled (SHG) and frequency triplicated (THG) in an frequency conversion stage, which emits radiation of 532 nm and 355 nm. The UV-radiation with 0,3 mJ of pulse energy is picked out and focused onto the sample for initiating the desorption process of the adsorbed molecules from the sample surface (fig. 3). The desorption is driven by the hot electronic excitation by the desorption laser and follows the theory of DIET-processes[3,4] from the MGR-modell by Menzel, Gomer and Redhead.
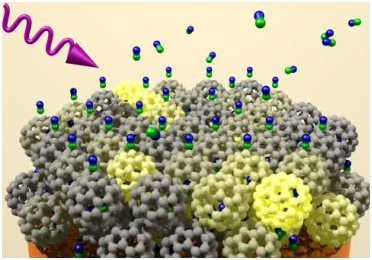
For the desorption experiment the primed sample is shot by a divergent NO-beam from a pulsed nozzle, so that the NO molecules physisorbing onto the sample. During this adsorption the NO molecules are creating (NO)2 dimers, where the oxygen atoms are aligned in “ortho” or in “para” position. By this dimer creation the number of adsorption positions is minored.
The ionization/detection laser system consists of a second Nd:YAG laser, which emits 130 mJ @ 532 nm pulsed radiation and 15 mJ @ 355 nm after frequency doubling and triplicating. The 532 nm radiation pumps a PDL-2 Dye Laser, which is driven emits tunable radiation of 13 mJ @ ~630 nm.
Both the 355 nm and the tunable 630 nm beams are frequency mixed in a BBO-Autotracker to get tunable UV-radiation in the range of 224 - 227 nm with a pulse energy of 0,7 mJ .
This frequency mixed beam is focused into the opening slit of the detector to ionize the desorbed molecules from the surface. The ionization of the molecules follows the process of the so called (1 +1)-REMPI–process (Resonantly Enhanced Multi Photon Ionization), where the first photon excites the Molecule in a higher state and by the second photon the molecule gets ionized. The Ionization energy then depends on the rotational and vibrational (=”rovibrational”) state of the molecule, so for detecting the different “rovibrational” state the laser energy has to be passed through.
In the beginning the C60 /Cu(111) sample has to be primed by a definite procedure of alternating evaporating C60 –molecules onto the Cu(111) sample. The preparation of the sample is observed elementary by XPS (fig. 4, left) and its electronic states are determined by UPS (fig. 4 right). After a preparation time of 20hours there is no Copper peak visible but a clear carbon peak in the XPS spectrum and the electronic states of C60 in the UPS spectrum visible. So the Cu(111) sample is completely covered by several monolayers.

Experimental Results
For each rotational state a corresponding time-of-flight or a drift-time distribution is measured and transformed into a velocity distribution, as shown in figure 4.
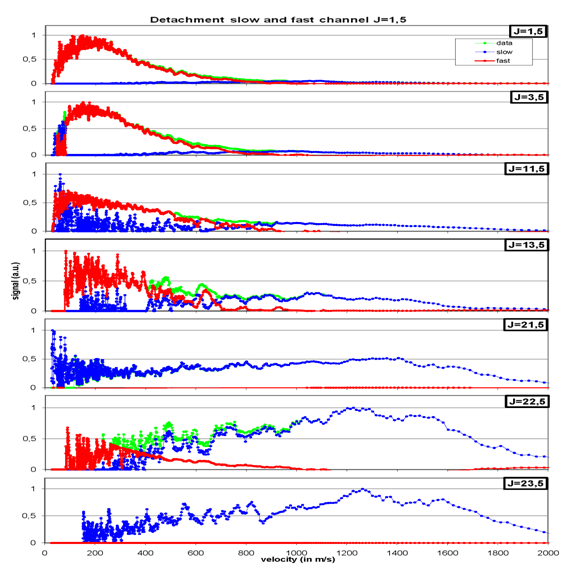
By fitting a “Boltzmann-Plot” into the time-of-flight distributions one can in the corresponding velocity distributions (fig. 5), that the molecules are desorbing with two different “velocity channels”. One the one hand there are fast molecules (blue line), who’s signal rises with rising rotational state, on the other hand we observed molecules with slower velocities (red line). In the case of the slow molecules it was found, that desorption occurs after a short time delay and their desorbing velocity is quite high like the fast molecules. Further analysis concerning variations of the surface temperature, the photon-flux onto the sample and the detection time delay lead to the founded assumption that the delayed desorption of t=127 µs might be induced by the long-living electronic excitations of the C60 –molecules (see fig. 1). So the molecules in the slow channel are not directly desorbing.
Also we could show that the higher the rovibrational state of the desorbing molecules, the higher is the velocity of the molecules. In other words: the translational energy of the NO-molecules rises with increasing rotational energy. So we could show a hard “translational-rotational coupling” in the energy of the desorbing molecules, as it is illustrated in fig. 5 by the dashed line. Among this one can see, that the number of the desorbing molecule in the slow channel gets lower with increasing rotational state up to J=23,5.
Outlook
- measurment of arrival time distributions at different rotational states
- measurement and analyzation of the results of the desorption for different vibrational states, as the states ν”=0, 1
- application of different desorption gases like Xe, CO, O2, NH3 in order to determine the behavior of d
- different small or diatomic air-molecules.
[1] H. W. Kroto, A. W. Allaf, S. P. Balm “C60: Buckminsterfullerene”, Chem. Rev., 1991, 91 (6) pp. 1213–1235
[2] Osawa, E. Superaromaticity. Kagaku (Chemistry) 1970, 25 854-863 (In Japanese.)
[3] J. A. Misewich, T. F. Heinz, D. M. newns, Phys. Rev. Lett. 68, 3737 (1992)
[4] D. Menzel, R. Gomer, J. Chem. Phys. 41, 331 (1964)