|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Free Neuropathology 6:3 (2025) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Original Paper |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Parkinsonism associated with prolonged unresponsive wakefulness syndrome after blunt head injury: a clinico-pathological study |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Kurt A. Jellinger |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Institute of Clinical Neurobiology, Vienna, Austria |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Corresponding author: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Submitted: 23 October 2024 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Keywords: Blunt traumatic brain injury, Prolonged unresponsive wakefulness, Chronic vegetative state, Posttraumatic parkinson-like symptoms, Brainstem lesions, Neuropathology |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Abstract
Objective: Survival after traumatic brain injury (TBI) and posttraumatic parkinsonian-like symptoms is increasing, in particular in those patients developing during disease course an unresponsive wakefulness syndrome (UWS) previously termed persistent vegetative state. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
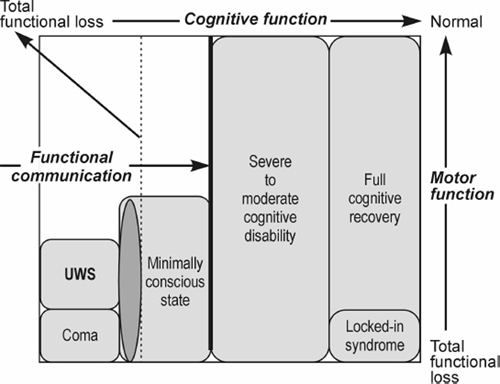
Introduction Survival rates after blunt traumatic brain injury (TBI) and chronic posttraumatic disorders of consciousness (DOC) are increasing [1]. In view of recent advances in the prognosis of TBI and related DOCs and their impact on diagnostic accuracy, prognosis and treatment [2, 3], the European Task Force on Disorders of Consciousness proposed the term "unresponsive wakefulness syndrome" (UWS) [4–8] for the challenging neurological conditions previously termed "apallic syndrome" [9, 10] or "persistent vegetative state" (PVS) [11, 12]. The functional outcomes following severe TBI are summarized in Fig. 1. With increased survival rates after severe TBI, the incidence of posttraumatic parkinsonism (PTP) has increased [14]. PTP is commonly caused by injuries from accidents (mainly motor vehicle accidents and falls) with loss of consciousness and latency to symptom onset of one to six or more months, although up to 42 % of patients did not report loss of consciousness [14]. In the fully developed "apallic syndrome", akinesia, amimia and increased muscle tonus with cogwheel phenomenon are almost obligatory and associated with spasticity and disorders of optomotoric akin to decerebration rigidity, while tremor is usually absent [10]. On the other hand, some patients with DOC following severe TBI have been reported to have parkinsonian symptoms [15]. Both PVS and minimally conscious state (MCS) following severe TBI can include features of akinetic mutism and parkinsonism that may share midbrain network dysfunctions [16, 17]. Extrapyramidal symptoms with bradykinesia, rigidity and resting tremor are the hallmark features of PTP, whereas postural instability was reported only in 15 % of cases [18].
Figure 1. Neuroimaging shows either a subdural hematoma causing transient compression obstruction of the basal ganglia or hypointense lesions confined to the basal ganglia [14] or were negative [15]. In a prospective study of patients with posttraumatic movement disorders, basal ganglia were the most common sites affected (66.7 %), followed by thalamus and brainstem (16.7 % each), while diffuse white matter (WM) involvement was the most common radiological lesion in patients with tremor [19]. In a man aged 37 years, parkinsonian features following a blunt TBI were associated with hypodense computer tomography (CT) lesions in the substantia nigra (SN) region [20]. In three patients, a posttraumatic rigid-akinetic syndrome resembling Parkinson disease appeared after a delay of one to five months after TBI. These patients displayed traumatic changes in the substantia nigra (SN) according to neuroimaging, and responded to levodopa [21]. According to a recent review, 53 % of PTP cases had a confirmed lesion; 53 % of the latter was located in the SN, 22 % more broadly in the midbrain [14]. Neuropathological studies of PTP after blunt TBI emphasized the importance of brainstem lesions secondary to increased intracranial pressure (ICP) [22–25] and extensive damage of the cerebral WM due to diffuse axonal injury [26–28], associated with hippocampal lesions and bilateral necrosis in thalamus and other basal ganglia [29]. Among 35 patients who survived blunt TBI for one month and up to eight years, thalamic lesions were seen in up to 96 % of cases, while diffuse axonal injury and ischemic brain lesions were seen in 14 % to 71 % of cases [30]. Brain-injured patients with DOC showed lesions of thalamus and basal nuclei that were confirmed by MRI [31] or proton magnetic spectroscopy [32]. Thalamic lesions were associated with thalamo-cortical disconnectivity [33] and disordered brain networks [34, 35], whereas brainstem lesions after severe TBI were of prognostic value [36]. Disruption of the ascending arousal network in the brainstem tegmentum causes DOC after acute TBI [37]. Neuroimaging studies suggested an important role for the default mode network to discriminate the UWS from MCS [8, 38], while abnormalities in the brainstem were confined to the TBI group [39]. Brainstem injuries detected by MRI after severe TBI were shown to be of prognostic value [36]. Most recent studies provided evidence for a key difference in the left frontoparietal connectivity when contrasting UWS with MCS [40]. This paper presents the neuropathological findings in a series of 15 patients surviving after blunt TBI, showing either complete recovery or defective states of UWS, presenting with severe to mild, mainly symmetrical rigid-akinetic parkinsonian symptoms. Material and methods In a consecutive post-mortem series of 630 subjects who survived a blunt TBI, 100 subjects (59 males and 41 females, age from five to 85 years, mean age 41.5 years, 77 % traffic accidents, 23 % falls plus other causes) with prolonged DOC and a survival between 12 and 900 days showed various states of consciousness ranging from deep coma or acute midbrain syndrome (n = 6, survival 12–35 days) to full UWS state (n = 61, survival 12–257 days), partial recovery from UWS (n = 22, survival 21–900 days) to remission state or partial clinical improvement comparable to MCS (n = 11, survival 21–293 days, mean 93.5 days). The neuropathological findings of this group have been described previously [25]. The 100 patients who survived blunt TBI for periods between 22 and 900 days (mean 564 days) included 15 patients (13 males, 2 females aged 21 to 56 years, mean 41.4 years) who showed not only posttraumatic motor and postural disorders but also flexor and / or extensor spasms and optomotor disorders (diplopia, convergence disorders) and who developed parkinson-like symptoms of moderate to severe intensity, mainly characterized by symmetrical hypo / akinesia, hypomimia / aminia and rigidity, with convergence disorders in 10 patients and unilateral or bilateral resting tremor in six patients. The clinical data of these 15 patients were evaluated retrospectively from their clinical and intensive care records. Their state of consciousness was evaluated by intensive care specialists according to the Glasgow scales and their extrapyramidal symptoms by movement disorder specialists. Since a part of the patients died between 1966 and 1980, CT studies could only be performed in a few patients. In addition to supratentorial traumatic lesions, the patients showed recent uni- or bilateral necroses, hemorrhagic cysts, old necroses, and/or atrophy of striatum, globus pallidus and/or thalamus, unilateral or bilateral hippocampal lesions, extensive damage to the cerebral WM and uni- or bilateral lesions in the brainstem, usually in lateral or dorsolateral parts of the pontine tegmentum, cerebral peduncles and/or cerebellum. In some patients, cerebral blood flow measurements were performed with the 133Xenon clearance method according to Ingvar & Lassen and gray matter hypoperfusion was correlated with the anatomical brainstem lesions [41]. Most of the patients with parkinsonian symptoms received levodopa treatment, initially per gastric tube and later on orally (levodopa / benserazide carbidopa 100 / 25 mg three times per day). Eleven patients showed partial improvement of the parkinsonian symptoms and less PVS, while four patients showed almost complete recovery of both syndromes. Neuropathological examination was performed according to standard protocols, with macroscopic assessment of brain atrophy, focal supratentorial brain lesions, deep traumatic and posttraumatic brain lesions (residuals of subdural / epidural or subarachnoidal hemorrhages, cortical contusions / concussions or hematomas, etc.). Pressure necroses / hemorrhages in the parahippocampal gyri were taken as evidence that ICP had been increased. Macroscopic changes in brainstem and cerebellum were described in detail. Histological examination of multiple paraffin blocks was performed using routine stains (H&E, cresyl violett, Kluver-Barrera or Heidenhain stain, Bodian silver impregnation, Holzer stain for fibrillary gliosis and immunohistochemistry for glial fibrillary acidic protein / GFAP). Amyloid-β, tau and synuclein pathologies were not specifically examined. Results The type, location and extension of the essential morphological changes are presented in Table 1. The key clinical and neuropathological data are presented in Table 2.
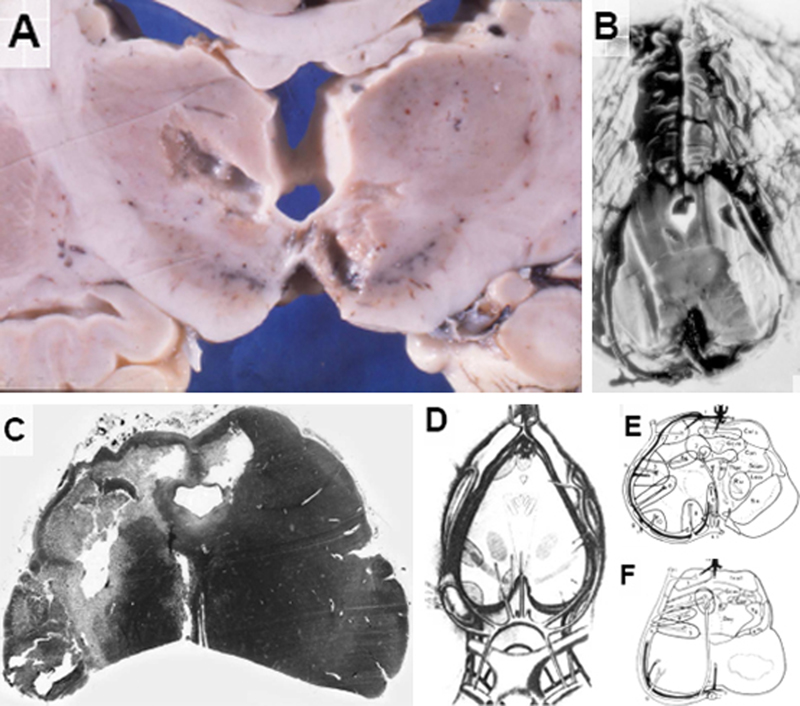
The 10 patients who survived for periods between 25 and 900 days with a partial recovery from UWS showed residuals of intracranial epi- or subdural hemorrhages (n = 4), cortical contusions / lacerations (n = 8), vascular / ischemic cortical necroses or WM hemorrhages (n = 1 each), diffuse WM changes (n = 6), lesions of the corpus callosum (n = 1), old hemorrhagic / ischemic lesions of the hippocampus usually with unilateral predominance (n = 9) or old hemorrhages and / or (cystic) necroses of the globus pallidus (n = 5) or less frequently of the striatum and / or the thalamus (n = 3/2). Among the latter patients, one case each also showed cystic necrosis of the globus pallidus with unilateral or bilateral lesions (small cysts, old hemorrhages, neuronal depletion) in the SN. The lesions mainly involved the lateral and dorsal parts of the SN (supply areas of small circumflexious rami) or oral and medial parts of the SN (supply areas of paramadian arteries). No definite lesions were seen around the walls of the third ventricle and in the periventricular gray matter. In one case, small cystic necroses and hemorrhagic residuals were detected in the dorsolateral midbrain tegmentum together with bilateral SN lesions. All the other cases showed superficial defects or hemorrhagic residuals in the lateral and dorsolateral pontine tegmentum. In one case each, myelin pallor and diffuse gliosis of the pontine basis and cerebellar lesions were present. A 32-year-old woman victim of a skiing accident was treated by excavation of an epidural hematoma. She survived for 10 days in an acute coma and for 257 days in partial recovery from UWS. She had no further direct traumatic brain lesions. In addition to spastic hemiparesis, she developed symmetric rigidity, akinesia and amimia, but reacted to external stimuli and, finally spoke a few hardly understandable words. She died from pneumonia. Neuropathology revealed residuals of the epidural hematoma but no primary traumatic lesions, subarachnoid hemorrhage or cortical contusions. Yet, there were cystic necroses of the left central thalamus and contralateral medial hypothalamus next to the SN (Fig. 2A), compression necrosis of the left hippocampus/parahippocampus (not shown) and symmetric cystic necroses in the caudal midbrain colliculi (Fig. 2B) and extensive unilateral necrosis of the SN and pes pedunculi (Fig. 2C). The location of the most frequent brainstem lesions due to transtentorial shifting and compression as well as the corresponding arterial blood supply are shown in Fig. 2 D–F.
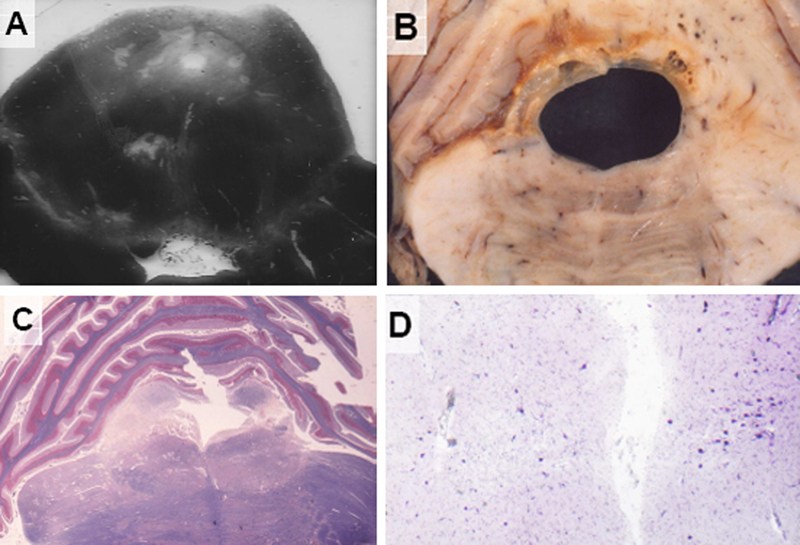
Figure 2. A 39-year-old man sustained a drunken fall on his occiput. No skull fracture, intracerebral hemorrhages or cortical contusions were found. Following a deep coma lasting four days, he died after 456 days in UWS with moderate clinical improvement, spasticity of his extremities, double vision (paresis of the right third brain nerve), bilateral rigidity, akinesia and hypomimia without tremors. Cerebral blood flow measurement at 100 days post TBI showed considerable reduction of gray matter perfusion, which significantly correlated with the anatomical brainstem lesion. After about 6 months, he was extubated, and later began to speak single words and to react to commands. CT showed cystic lesions in bilateral basal ganglia and dorsolateral brainstem. Levodopa was administered first through gastric tube and later on orally with levodopa / benserazide 100 + 25 mg three times per day. Levodopa treatment led to slow reduction of rigidity and amimia but did not change spasticity. The patient was transferred to a chronic care center, where his general state did not change considerably. He died from acute cardiac infarction. Neuropathology revealed no superficial traumatic residuals but diffuse WM lesions and old cystic necroses in globus pallidus and thalamus due to compression of the left anterior choroidal and right thalamo-perforant arteries. In addition to an old hemorrhagic infarction in the right occipital lobe, extensive lesions were found in the hippocampus/parahippocampus with unilateral predominance. There were small cystic necroses in the left dorsolateral midbrain tegmentum around the aqueduct, in the nucleus ruber and in the ipsilateral SN (Fig. 3A). Old hemorrhagic compression necroses in the pontine tegmentum surrounded the enlarged aqueduct (Fig. 3B), while extensive necroses were found in the pontine tegmentum affecting the rostral reticular formation and central tegmental tract (Fig. 3C). In the SN, small cystic lesions and focal loss of neurons were seen (Fig. 3D).
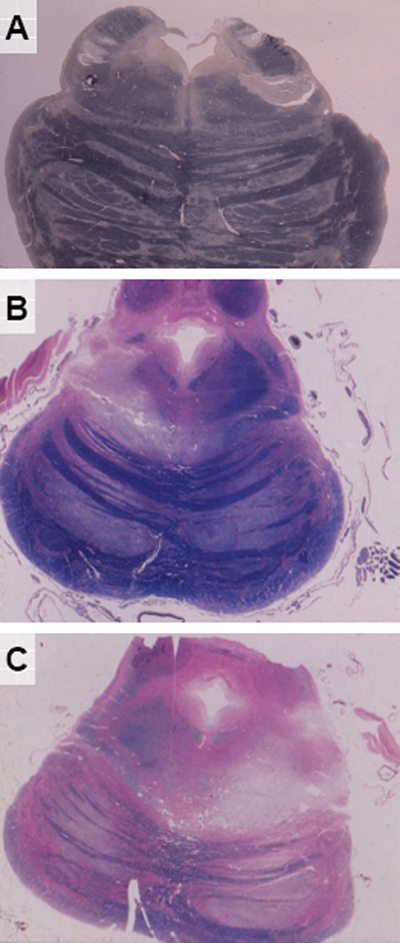
Figure 3. A 36-year-old motorcyclist, hit by a car, received a blow to the right forehead, causing no skull fracture, intracranial hematoma or cortical contusions. He was in deep coma for around 30 days and died after 301 days, transition from UWS to Klüver-Bucy syndrome. In addition to left-sided spastic hemiparesis, he developed parkinsonian symptoms with almost bilateral akinesia, rigidity, hypomimia and slurred, hardly understandable speech. He was given levodopa via gastric tube and later on orally, which was followed by incomplete reduction of rigidity and akinesia without changes of spastic hemiparesis. He died from pneumonia. Neuropathology revealed neither skull fracture nor residuals of intracranial hemorrhage or cortical contusions, but diffuse WM changes, partial rupture of corpus callosum and severe hippocampal/parahippocampal lesions with left preponderance. The basal ganglia, walls and bottom of the third ventricle as wells as the periventricular gray matter and oral midbrain were preserved. Old hemorrhagic necroses involved however the dorsal and dorsolateral tegmentum (Fig. 4A and C). A small necrosis was seen in the right dorsolateral pontine tegmentum (Fig. 4B), while extensive compression necroses were seen in the right dorsolateral pontine tegmentum and adjacent cerebellar gyri (Fig. 4D). Pictures of the bilateral SN lesions have unfortunately been lost.
Figure 4. Five subjects survived TBI for periods ranging from 22 to 293 days in various recovery states from UWS, presenting with MCS associated with spasticity and moderate to mild parkinsonian symptoms. Neuropathology revealed residuals of subdural hemorrhage (n = 1) and cortical contusions (n = 2), callosal damage (n = 1) and diffuse WM lesions (n = 3), uni- or bilateral lesions of globus pallidus and/or thalamus with or without lesions to the SN were seen in one case each, as well as hippocampal/parahippocampal lesions (n = 2). Brainstem lesions were restricted to old uni- or bilateral necroses in dorsolateral pontine tegmentum (Fig. 5A) that were associated with diffuse pontine gliosis in one case. A 30 year-old man, a motorcyclist hit by a car, survived after evacuation of a subdural hematoma for 301 days, first 20 days in deep coma and subsequently with moderate improvement in UWS. In addition to right-sided spasticity, he developed almost symmetrical akinesia, bilateral rigidity and hypomimia with mild unilateral resting tremor. He died in 1968 hence no MRI was possible. He also received levodopa through gastric tube and later orally, which was followed by reduction of rigidity and hypomima without changes of spasticity. He died from pneumonia. Neuropathology revealed a brain fracture and residuals of subdural hematoma and superficial cortical contusions as well as bilateral partial necrosis of striatum and globus pallidus, diffuse WM lesions and considerable hippocampal lesions with left-sided predominance. The floor of the 3rd ventricle, the periaqueductal gray matter and the midbrain tegmentum were relatively preserved, while old necroses involved the dorsal and dorsolateral pontine tegmentum and the brachium conjunctivum (Fig. 5A). Another vascular necrosis in the dorsolateral tegmentum of the rostral pons (Fig. 5B) was associated with partial degeneration of the pyramidal tracts in the pontine basis (Fig. 5C).
Figure 5. Discussion Parkinsonian symptoms are well recognized as a component of posttraumatic encephalopathy in boxers and in other sport-related traumatic brain injuries (TBI) [42–44]. The incidence of parkinsonism after TBI (PTP) has been estimated to 3.07 % overall and to 11.59 % in TBI patients aged over 65 years [45]. Almost 100 years ago, the minimal criteria for the diagnosis of Parkinson's syndrome following acute head injury (PTP) were that (1) the trauma should be severe and should have led to concussion or unconsciousness, (2) there should be a close temporal relation between the acute trauma and the onset of parkinsonian features and the course of parkinsonian features should be uninterrupted [46]. The number of PTP cases after acute TBI in the earlier literature is limited [20, 47, 48]. A man aged 36 years with a skull fracture and unconsciousness for 24 hours developed over six weeks a predominantly right-sided, slowly progressive, levodopa-unresponsive parkinsonian syndrome. Neuroimaging disclosed a cerebral infarction in the left caudate and lenticular nucleus, suggesting impairment of nigro-striato-frontal circuits [49]. Other studies suggested that in PTP, supplementary motor area impairment seems important [50]. In a series of 54 patients who died in prolonged coma after closed TBI, a man aged 30 years died 301 days after a motorcycle accident in a partial recovery state from PVS, following a coma of 40 days. He had no skull fracture, cortical contusions or intracerebral hemorrhage but presented with parkinsonian features in addition to chronic decerebration. Autopsy revealed extensive brainstem lesions due to tentorial compression of the rostral brainstem as complications of increased ICP [22]. Matsuda and colleagues [51] reported three patients with PVS after severe TBI who, after recovering from prolonged loss of consciousness, presented parkinsonian features with mainly rigidity and hyperkinesia, which improved after levodopa treatment. T2-weighted MRI studies showed lesions in the dorsolateral midbrain and the cerebral peduncle, suggesting axonal injury involving the dopaminergic system (SN and ventral tegmental area). These findings implied that the midbrain was injured by tentorial compression induced by translatory and rotatory acceleration when the cranium was struck in its sagittal axis, or by posterolateral damage [52, 53]. This conclusion was based on neuroradiological and neuropathological studies on PVS after TBI, indicating that the most common structural lesions were diffuse axonal injury involving the corpus callosum, the thalamus and the dorsolateral aspects of the rostral brainstem [26, 54]. A study using voxel-based morphometry on structural MRI involving 61 patients with DOC of traumatic origin found widespread structural brain injury in the brainstem, midbrain, thalamus, hypothalamus, basal forebrain, cerebellum, and posterior corpus callosum. Potential structural differences were found between UWS and MCS, especially at the individual level [7]. Primary traumatic brainstem lesions after severe TBI inducing parkinsonian symptoms are either caused by hyperextension of the head, tearing forces or rotation mechanisms leading to contusions, fatal tears and hemorrhages of the rostral brainstem [55] or by shearing motion due to a rotatory mechanism and cavitation forces inducing hemorrhages and strains in the upper brainstem [23]. This is documented by two personal observations: first, a 19-year-old motorcyclist who died after 18 hours in deep coma after a rear-end collision crashing into a tree showed rupture of the diencephalon and hemorrhages in the SN; second, a 37-year-old man who survived 10 days in deep coma after a drunken fall with multiple fractures of the calvaria showed multiple cortical lacerations, deep diencephalic ruptures and hemorrhages in midbrain SN and rostral pons [56]. Rare cases of patients with traumatic SN lesions who survived for a longer time with or without developing a parkinsonian syndrome have been reported, such as a subject who survived a severe TBI with symmetric SN necroses for 32 years without parkinsonism [57]. The findings in the present study of patients who developed parkinsonian symptoms in the course of long-term UWS after blunt TBI and in an earlier study of a larger group of patients with prolonged UWS after severe TBI [25] are in favor of a secondary traumatic cause of morphological lesions related to prolonged posttraumatic UWS, recovery from UWS and defective states including posttraumatic parkinsonian symptoms. This conclusion is based on the following facts: (1) Translatory and rotational acceleration following TBI is usually followed by extensive cortical contusions/lacerations at the contralateral side of the focal impact, often associated with lesions or rupture of the corpus callosum, which was seen in only 13.3 % of cases in the present series. (2) Direct or "primary traumatic" lesions to the rostral or caudal brainstem due to rotational shearing injury or rotatory acceleration forces are usually associated with acute death [56, 58, 59]. However, in rare patients with long survival following acute TBI, symmetrical necrosis of the SN without clinical parkinsonian symptoms has been reported [57]. In the majority of cases with prolonged survival in UWS and recovery states, the morphological lesions are restricted to the dorsal and dorsolateral pontine tegmentum, with preservation of the upper brainstem. (3) When comparing the frequency of brainstem lesions and morphological signs of increased ICP, there is strong association between both parameters. This is particularly true for the UWS group, when considering the presence of intracranial expanding lesions, the history of elevated ICP and the morphological sequelae of elevated ICP in various cerebral regions, e.g., compression necrosis in the hippocampus, which is seen in up to 80 % of fatal TBIs [25, 60–62]. Patients surviving TBI in deep coma, in addition to frequently showing both intracranial hemorrhages, cortical contusions (about 80 %) or hippocampal lesions (only 50 %), showed multiple brainstem lesions mostly involving the floor of the 3rd ventricle, the periaqueductal gray matter and the brainstem tegmentum, involving the ascending arousal system (AAS) or the ascending arousal network (AAN) [29, 37, 58, 63]. These lesions are frequently associated with diffuse axonal injury [28, 64, 65], disrupting the connection between brainstem, thalamus, forebrain, and substantially impairing the activation of the default mode network [66], the resting state and other functional networks [67]. While some patients will irreversibly remain in UWS, particularly due to severe damage to central areas of the upper brainstem, usually involving sequelae of increased ICP and transtentorial compression of the upper brainstem, other patients evolve to a minimally responsive condition or MCS [68, 69]. In these cases, brainstem lesions are usually restricted to dorsolateral/peripheral parts of the pontine tegmentum with preservation of the AAS [17]. As illustrated in a former and in the present case series, these patients, irrespective of occasionally frequent supratentorial lesions, may show involvement of the dorsolateral pontine tegmentum, the cerebral peduncles and the SN, with preservation of the central parts of the rostral brainstem. There is evidence that during recovery from traumatic coma, there is increase in brainstem-thalamic or AAN connectivity [70]. The distribution pattern of the brainstem lesions in the present series of posttraumatic UWS and residual parkinsonian symptoms, i.e., superficial softening in the dorsolateral tegmentum of the rostral pons and the velum medullare anterior, corresponds to the supply area of branches of the long lateral midbrain and pontine vessels, the superior cerebellar and posterior perforating arteries or of the basal and internal cerebral veins. Focal lesions in the dorsolateral pontine tegmentum affecting the superior cerebellar peduncle and the lateral lemniscus correspond to the supply areas of branches of long circumferential and superior cerebellar arteries or the drainage territory of precentral cerebellar veins as well as of the lateral sulcus veins. Occasional small scattered necroses and hemorrhagic scars in the pontine tegmentum result from obstruction of branches of circumferential vessels or the superior cerebellar artery (Fig. 2 D–F). These circulatory disorders result from compression of the brainstem tegmentum, leading from increased ICP to free tentorial edge / transtentorial shifting. The lesion pattern of posttraumatic parkinsonian symptoms and the therapeutic efficacy of long-term levodopa treatment suggest a dysfunction of the nigro-striato-cortical catecholaminergic system [14, 71], which may, however, progressively decompensate in some patients with long-lasting UWS after severe TBI. The limitations of the present study based on archival material are threefold: the absence of MRI data and / or SPECT and PET data on striatonigral dysfunction [16], the lack of immunohistochemical data on Alzheimer-related lesions, in particular of analyses of p-tau and β-amyloid to exclude changes akin to chronic traumatic encephalopathy [72, 73] and the lack of data on amyloid precursor protein to detect axonal injury [74], which was partly detected in the present study by Bodian silver impregnation. Another limitation is the fact that disorders previously diagnosed as "apallic syndrome" or PVS had to be reclassified using current criteria (UWS). The essential point of the present study is to demonstrate the morphological basis of rare parkinsonian symptoms in the course of UWS following fatal TBS. This endeavour appears important for further clinical and prognostic studies aiming to improve assessment of brain (dys)functions in prolonged DOC / UWS. Here, modern neuroimaging and other markers may provide valuable signs, not only documenting changing nosology but also offering further insights into the pathophysiology of posttraumatic parkinsonism. Acknowledgements The author thanks Mr. E. Mitter-Ferstl for secretarial and editorial work. Funding Statement The study was funded by the Society for the Promotion of Research in Experimental Neurology, Vienna, Austria. Conflict of Interest Statement The author declares that he has no conflict of interest. Ethics approval This is a retrospective analysis of archival material of anonymized patient data from the years 1958–1980, for which no later ethical approval could be achieved. Abbreviations CT - computer tomography, DOC - disorder of consciousness, ICP - intracranial pressure, MCS - minimally conscious state, SN - substantia nigra, PTP - posttraumatic parkinsonism, PVS - persistent vegetative state, TBI - traumatic brain injury, UWS - unresponsive wakefulness syndrome, WM - white matter. References 1. Yan D, et al. (2024) Prognosis of patients with prolonged disorders of consciousness after brain injury: a longitudinal cohort study. Front Public Health 12:1421779. https://doi.org/10.3389/fpubh.2024.1421779 2. Schnakers C, et al. (2009) Diagnostic accuracy of the vegetative and minimally conscious state: clinical consensus versus standardized neurobehavioral assessment. BMC Neurol 9:35. https://doi.org/10.1186/1471-2377-9-35 3. Maas AIR, et al. (2022) Traumatic brain injury: progress and challenges in prevention, clinical care, and research. Lancet Neurol 21:1004-60. https://doi.org/10.1016/S1474-4422(22)00309-X 4. Laureys S, et al. (2010) Unresponsive wakefulness syndrome: a new name for the vegetative state or apallic syndrome. BMC Med 8:68. https://doi.org/10.1186/1741-7015-8-68 5. Bruno MA, et al. (2011) From unresponsive wakefulness to minimally conscious PLUS and functional locked-in syndromes: recent advances in our understanding of disorders of consciousness. J Neurol 258:1373-84. https://doi.org/10.1007/s00415-011-6114-x 6. van Erp WS, et al. (2014) The vegetative state/unresponsive wakefulness syndrome: a systematic review of prevalence studies. Eur J Neurol 21:1361-8. https://doi.org/10.1111/ene.12483 7. Guldenmund P, et al. (2016) Structural brain injury in patients with disorders of consciousness: A voxel-based morphometry study. Brain Inj 30:343-52. https://doi.org/10.3109/02699052.2015.1118765 8. Rosazza C, et al. (2016) Multimodal study of default-mode network integrity in disorders of consciousness. Ann Neurol 79:841-53. https://doi.org/10.1002/ana.24634 9. Kretschmer E (1940) Das apallische Syndrom. Z ges Neurol Psychiat 169:576-9. https://doi.org/10.1007/BF02871384 10. Gerstenbrand P (1967) Das traumatische apallische Syndrom. Wien: Springer. https://doi.org/10.1007/978-3-7091-8167-6 11. von Wild K, et al. (2012) The vegetative state - a syndrome in search of a name. J Med Life 5:3-15. PMID: 22574081 12. Jennett B, Plum F (1972) Persistent vegetative state after brain damage. A syndrome in search of a name. Lancet 1:734-7. https://doi.org/10.1016/S0140-6736(72)90242-5 13. Posner JB, et al. (2007) Plum and Posner's Diagnosis of Stupor and Coma, 4th Edition. New York: Oxford Univ. Press. https://doi.org/10.1093/med/9780195321319.001.0001 14. Rojvirat C, et al. (2024) Systematic review of post-traumatic parkinsonism, an emerging parkinsonian disorder among survivors of traumatic brain injury. Neurotrauma Rep 5:37-49. https://doi.org/10.1089/neur.2023.0104 15. Takeda M, et al. (1991) A case of posttraumatic parkinsonism. Rinsho Shinkeigaku 31:842-6. PMID: 1764860 16. Jenkins PO, et al. (2020) Distinct dopaminergic abnormalities in traumatic brain injury and Parkinson's disease. J Neurol Neurosurg Psychiatry 91:631-7. https://doi.org/10.1136/jnnp-2019-321759 17. Rosenblum WI (2015) Immediate, irreversible, posttraumatic coma: a review indicating that bilateral brainstem injury rather than widespread hemispheric damage is essential for its production. J Neuropathol Exp Neurol 74:198-202. https://doi.org/10.1097/nen.0000000000000170 18. Formisano R, Zasler ND (2014) Posttraumatic parkinsonism. J Head Trauma Rehabil 29:387-90. https://doi.org/10.1097/htr.0000000000000027 19. Manjunath N, et al. (2019) Posttraumatic movement disorders: A clinical and imaging profile of 30 patients. Neurol India 67:738-43. https://doi.org/10.4103/0028-3886.263212 20. Nayernouri T (1985) Posttraumatic parkinsonism. Surg Neurol 24:263-4. https://doi.org/10.1016/0090-3019(85)90035-7 21. Bhatt M, et al. (2000) Posttraumatic akinetic-rigid syndrome resembling Parkinson's disease: a report on three patients. Mov Disord 15:313-7. https://doi.org/10.1002/1531-8257(200003)15:2<313::aid-mds1017>3.0.co;2-p 22. Jellinger K, Seitelberger F (1970) Protracted post-traumatic encephalopathy. Pathology, pathogenesis and clinical implications. J Neurol Sci 10:51-94. https://doi.org/10.1016/0022-510X(70)90091-2 23. Mayer ET (1967) [Central brain stem lesions after the effect of blunt violence to the skull. Brain stem lesions]. Arch Psychiatr Nervenkr (1970) 210:238-62. https://doi.org/10.1007/BF00367551 24. Mayer ET (1968) [Clinical and pathological aspects of the traumatic mesencephalic and apallic syndrome]. Arztl Forsch 22:163-72. PMID: 5755585 25. Jellinger KA (2013) Neuropathology of prolonged unresponsive wakefulness syndrome after blunt head injury: review of 100 post-mortem cases. Brain Inj 27:917-23. https://doi.org/10.3109/02699052.2013.793395 26. Adams JH, et al. (2000) The neuropathology of the vegetative state after an acute brain insult. Brain 123 (Pt 7):1327-38. https://doi.org/10.1093/brain/123.7.1327 27. Graham NSN, et al. (2020) Diffuse axonal injury predicts neurodegeneration after moderate-severe traumatic brain injury. Brain 143:3685-98. https://doi.org/10.1093/brain/awaa316 28. Strich SJ (1956) Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry 19:163-85. https://doi.org/10.1136/jnnp.19.3.163 29. Graham DI, et al. (2005) Novel aspects of the neuropathology of the vegetative state after blunt head injury. Prog Brain Res 150:445-55. https://doi.org/10.1016/s0079-6123(05)50031-1 30. Jennett B, et al. (2001) Neuropathology in vegetative and severely disabled patients after head injury. Neurology 56:486-90. https://doi.org/10.1212/wnl.56.4.486 31. Lutkenhoff ES, et al. (2015) Thalamic and extrathalamic mechanisms of consciousness after severe brain injury. Ann Neurol 78:68-76. https://doi.org/10.1002/ana.24423 32. Uzan M, et al. (2003) Thalamic proton magnetic resonance spectroscopy in vegetative state induced by traumatic brain injury. J Neurol Neurosurg Psychiatry 74:33-8. https://doi.org/10.1136/jnnp.74.1.33 33. Laouchedi M, et al. (2015) Deafferentation in thalamic and pontine areas in severe traumatic brain injury. J Neuroradiol 42:202-11. https://doi.org/10.1016/j.neurad.2014.03.001 34. Zhou J, et al. (2011) Specific and nonspecific thalamocortical functional connectivity in normal and vegetative states. Conscious Cogn 20:257-68. https://doi.org/10.1016/j.concog.2010.08.003 35. Vanhaudenhuyse A, et al. (2010) Default network connectivity reflects the level of consciousness in non-communicative brain-damaged patients. Brain 133:161-71. https://doi.org/10.1093/brain/awp313 36. Hilario A, et al. (2012) Severe traumatic head injury: prognostic value of brain stem injuries detected at MRI. AJNR Am J Neuroradiol 33:1925-31. https://doi.org/10.3174/ajnr.a3092 37. Snider SB, et al. (2019) Disruption of the ascending arousal network in acute traumatic disorders of consciousness. Neurology 93:e1281-e7. https://doi.org/10.1212/wnl.0000000000008163 38. Kim YW, et al. (2010) Voxel-based statistical analysis of cerebral glucose metabolism in patients with permanent vegetative state after acquired brain injury. Chin Med J (Engl) 123:2853-7. PMID: 21034596 39. Newcombe VF, et al. (2010) Aetiological differences in neuroanatomy of the vegetative state: insights from diffusion tensor imaging and functional implications. J Neurol Neurosurg Psychiatry 81:552-61. https://doi.org/10.1136/jnnp.2009.196246 40. Ihalainen R, et al. (2024) Lateral frontoparietal effective connectivity differentiates and predicts state of consciousness in a cohort of patients with traumatic disorders of consciousness. PLoS One 19:e0298110. https://doi.org/10.1101/2023.06.07.544105 41. Heiss WD, Jellinger K (1972) Cerebral blood flow and brain stem lesion. Z Neurol 203:197-209. https://doi.org/10.1007/bf00316111 42. Critchley M (1957) Medical aspects of boxing, particularly from a neurological standpoint. Br Med J 1:357-62. PMID: 13396257 43. Jordan BD (2013) The clinical spectrum of sport-related traumatic brain injury. Nat Rev Neurol 9:222-30. https://doi.org/10.1038/nrneurol.2013.33 44. Krauss JK (2015) Movement disorders secondary to craniocerebral trauma. Handb Clin Neurol 128:475-96. https://doi.org/10.1016/b978-0-444-63521-1.00030-3 45. Harnod D, et al. (2019) Posttraumatic parkinsonism would increase the mortality risk in elderly patients with traumatic brain injury. Ann Transl Med 7:734. https://doi.org/10.21037/atm.2019.12.04 46. Crouson O, Justin-Besancon L (1929) Le Parkinsonism traumatique. Presse Med 37:1325-7. https://doi.org/10.1016/j.jns.2024.123242 47. Lindenberg R (1964) [The mechanisms of damage to the substantia nigra in brain traumas and the problem of posttraumatic parkinsonism]. Dtsch Z Nervenheilkd 185:637-63. PMID: 14158296 48. Turjanski N, et al. (1997) Dopaminergic function in patients with posttraumatic parkinsonism: an 18F-dopa PET study. Neurology 49:183-9. https://doi.org/10.1212/wnl.49.1.183 49. Doder M, et al. (1999) Parkinson's syndrome after closed head injury: a single case report. J Neurol Neurosurg Psychiatry 66:380-5. https://doi.org/10.1136/jnnp.66.3.380 50. Péran P, et al. (2014) Supplementary motor area activation is impaired in severe traumatic brain injury parkinsonism. J Neurotrauma 31:642-8. https://doi.org/10.1089/neu.2013.3103 51. Matsuda W, et al. (2003) Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry 74:1571-3. https://doi.org/10.1136/jnnp.74.11.1571 52. Matsumura A, et al. (1993) Prediction of the Reversibility of the Brain Stem Dysfunction in Head Injury Patients: MRI and Auditory Brain Stem Response Study. In: Nakamura N, Hashimoto T, Yasue M (eds) Recent Advances in Neurotraumatology. Springer-Verlag Tokyo, pp 192-5. https://doi.org/10.1007/978-4-431-68231-8_43 53. Matsumura A, et al. (1998) Persistent vegetative state. J Neurosurg 89:895-6. https://doi.org/10.3171/jns.1998.89.5.0895 54. Kampfl A, et al. (1998) The persistent vegetative state after closed head injury: clinical and magnetic resonance imaging findings in 42 patients. J Neurosurg 88:809-16. https://doi.org/10.3171/jns.1998.88.5.0809 55. Lindenberg R, Freytag E (1970) Brainstem lesions characteristic of traumatic hyperextension of the head. Arch Pathol 90:509-15. PMID: 5485108 56. Kothbauer P, et al. (1977) Concerning the question of posttraumatic parkinsonian syndromes. In: Meeting of the Austrian Parkinsonian Society, Vienna, 1977, pp 1-6. Vienna: Roche-Fortbildungs-Service. 57. Huhn B, Jakob H (1970) [Traumatic brain stem lesions with long-term survival. Contribution to the pathology of substancia nigra and the pontine syndrome]. Nervenarzt 41:326-34. PMID: 5453704 58. Edlow BL, et al. (2012) Neuroanatomic connectivity of the human ascending arousal system critical to consciousness and its disorders. J Neuropathol Exp Neurol 71:531-46. https://doi.org/10.1097/nen.0b013e3182588293 59. Ravikanth R, Majumdar P (2022) Prognostic significance of magnetic resonance imaging in detecting diffuse axonal injuries: analysis of outcomes and review of literature. Neurol India 70:2371-7. https://doi.org/10.4103/0028-3886.364066 60. Jellinger KA (1983) Secondary brainstem involvement in blunt head injury. In: VILLANI R (ed) Advances in Neurotraumatology. Excerpta Medica I.C.S. Amsterdam-Oxford-Princeton, pp 58-66. 61. Adams JH, et al. (1999) The neuropathology of the vegetative state after head injury. J Clin Pathol 52:804-6. https://doi.org/10.1093/brain/123.7.1327 62. Adams JH (1992) Head injury. In: Adams JH, Duchen LW (eds) Greenfield's Neuropathology. Oxford Univ. Press Oxford, pp 106-52. https://doi.org/10.1111/j.1750-3639.1992.tb00699.x 63. Edlow BL, et al. (2013) Disconnection of the ascending arousal system in traumatic coma. J Neuropathol Exp Neurol 72:505-23. https://doi.org/10.1097/nen.0b013e3182945bf6 64. Bruggeman GF, et al. (2021) Traumatic axonal injury (TAI): definitions, pathophysiology and imaging-a narrative review. Acta Neurochir (Wien) 163:31-44. https://doi.org/10.1007/s00701-020-04594-1 65. Johnson VE, et al. (2013) Axonal pathology in traumatic brain injury. Exp Neurol 246:35-43. https://doi.org/10.1016/j.expneurol.2012.01.013 66. Threlkeld ZD, et al. (2018) Functional networks reemerge during recovery of consciousness after acute severe traumatic brain injury. Cortex 106:299-308. https://doi.org/10.1016/j.cortex.2018.05.004 67. Ordóñez-Rubiano EG, et al. (2024) Resting state networks in patients with acute disorders of consciousness after severe traumatic brain injury. Clin Neurol Neurosurg 242:108353. https://doi.org/10.1016/j.clineuro.2024.108353 68. Monti MM, et al. (2010) Willful modulation of brain activity in disorders of consciousness. N Engl J Med 362:579-89. https://doi.org/10.1056/NEJMoa0905370 69. Bagnato S, et al. (2013) Emerging from an unresponsive wakefulness syndrome: brain plasticity has to cross a threshold level. Neurosci Biobehav Rev 37:2721-36. https://doi.org/10.1016/j.neubiorev.2013.09.007 70. Snider SB, et al. (2020) Ascending arousal network connectivity during recovery from traumatic coma. Neuroimage Clin 28:102503. https://doi.org/10.1016/j.nicl.2020.102503 71. Wee IC, et al. (2024) Long-term impact of diffuse traumatic brain injury on neuroinflammation and catecholaminergic signaling: potential relevance for Parkinson's disease risk. Molecules 29:1470. https://doi.org/10.3390/molecules29071470 72. Inserra CJ, DeVrieze BW (2023) Chronic traumatic encephalopathy. In: StatPearls [Internet], 2023/08/07 Edition. StatPearls Publishing Treasure Island (FL). PMID: 29262155 73. McKee AC, et al. (2023) Chronic traumatic encephalopathy (CTE): criteria for neuropathological diagnosis and relationship to repetitive head impacts. Acta Neuropathol 145:371-94. https://doi.org/10.1007/s00401-023-02540-w 74. Krieg JL, et al. (2023) Identifying the phenotypes of diffuse axonal injury following traumatic brain injury. Brain Sci 13:1607. https://doi.org/10.3390/brainsci13111607
Copyright: © 2025 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||