|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Free Neuropathology 5:32 (2024) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Original Paper |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Adult glioblastoma with Lynch syndrome-associated mismatch repair deficiency forms a distinct high-risk molecular subgroup |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Maria-Magdalena Georgescu |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
NeuroMarkers, Houston, Texas, USA |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Corresponding author: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Additional resources and electronic supplementary material: supplementary material |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Submitted: 17 September 2024 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Keywords: Glioblastoma, MMR deficiency, Lynch syndrome, TP53, PIK3CA, Chromatin remodeling genes, Multinucleated giant cells, p53 immuno-histochemistry, Immune checkpoint inhibitors, Ipilimumab, Nivolumab |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Abstract Glioblastoma is the most frequent and malignant primary brain tumor. Although the survival is generally dismal for glioblastoma patients, risk stratification and the identification of high-risk subgroups is important for prompt and aggressive management. The G1–G7 molecular subgroup classification based on the MAPK pathway activation has offered for the first time a non-redundant, all-inclusive classification of adult glioblastoma. Five patients from the large, 218-patient, prospective cohort showed germline mutations in mismatch repair (MMR) genes (Lynch syndrome) and a significantly worse median survival of 3.25 months post-surgery than those from the G1/EGFR and G3/NF1 major subgroups, or from the rest of the cohort adjusted for age. These rare tumors were assigned to a new subgroup, G3/MMR, a G3/NF1 subgroup spin-off, as they generally show genomic alterations leading to RAS activation, such as NF1 and PTPN11 mutations. An integrated clinical, histologic and molecular analysis of the G3/MMR tumors showed distinct characteristics as compared to other glioblastomas, including those with iatrogenic high tumor mutation burden (TMB), warranting a separate subgroup. Prior history of cancer, midline location or multifocality, presence of multinucleated giant cells (MGCs), positive p53 and MMR immunohistochemistry, and specific molecular characteristics, including high TMB, MSH2/MSH6 alterations, biallelic TP53 Arg mutations and co-occurring PIK3CA p.R88Q and PTEN alterations, alert to this high-risk G3/MMR subgroup. The MGCs and p53 immunohistochemistry analysis in G1–G7 subgroups showed that one in 7 tumors with these characteristics is a G3/MMR glioblastoma. The FDA-approved first-line therapy for many advanced solid tumors consists of nivolumab-ipilimumab immune checkpoint inhibitors. One G3/MMR patient received this regimen and survived much longer than the rest, setting a proof-of-principle example for the treatment of these very aggressive G3/MMR glioblastomas. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
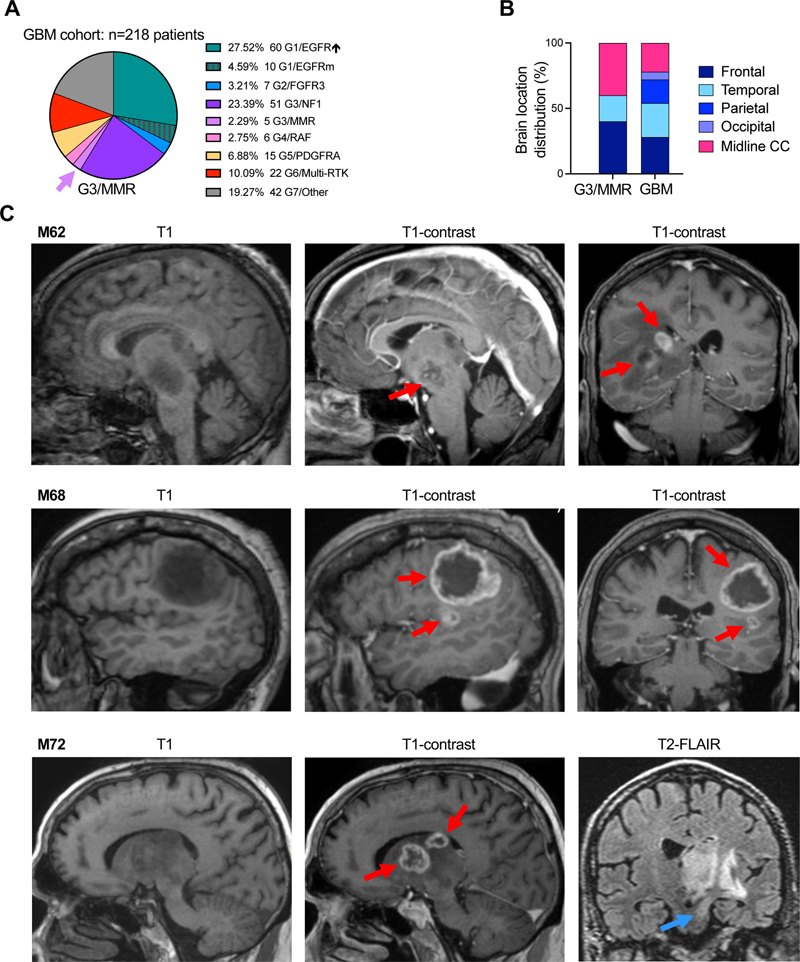
Introduction Glioblastoma is the most frequent and deadly primary brain tumor [1], assigned by the World Health Organization (WHO) Classification of Central Nervous System Tumors the highest tumor grade, WHO grade 4 [2]. Its dismal prognosis is mainly due to diffuse brain invasion, tumor heterogeneity, but also activation of pathways that impart resistance to therapy. Virtually all glioblastomas show activation of the extracellular signal-regulated kinase / mitogen-activated protein kinase (ERK/MAPK) and phosphatidyl-inositol 3-OH kinase (PI3K) canonical growth pathways [3–5]. Non-redundant molecular alterations in the upstream mediators of the ERK/MAPK pathway, including the receptor tyrosine kinases (RTKs) triggering pathway activation, allowed an all-inclusive molecular classification of glioblastoma into 7 subgroups [4,5]. These subgroups, termed after the most upstream gene showing activating RTK or MAPK effector alterations are: G1/EGFR, G2/FGFR3, G3/NF1, G4/RAF, G5/PDGFRA, G6/Multi-RTK, and G7/Other, the latter showing dominant PI3K pathway activation [4,5]. Of these, the G1/EGFR, G3/NF1 and G7/Other are major subgroups, containing together approximately three quarters of all glioblastoma cases, and showing distinct risk stratification, with overall poor prognosis for the G7/Other subgroup [5]. The treatment of glioblastoma relies on surgical resection, the extent of which correlates to the survival benefit [6]. Subsequent radiation and temozolomide (TMZ) chemotherapy represent the standard adjuvant regimen for the past 20 years [7], with few recent trials proven beneficial for the younger glioblastoma patient population [8]. Beside risk stratification, the aim of the G1–G7 molecular subgroup classification is to identify subgroups benefiting of specific targeted therapy [5]. For example, the G6/Multi-RTK has been analyzed in detail and shows RTK gene fusions for which addition of specific RTK inhibitors improves survival [5,9,10]. A very small but significant subgroup of glioblastomas that harbor germline mismatch repair (MMR) gene deficiency consistent with Lynch syndrome has been identified in a previous study [5]. This subgroup has been termed G3/MMR, as RAS activation appears to be dominant in these tumors, similarly to the G3/NF1 tumors. Lynch syndrome is a cancer predisposition syndrome caused by germline pathogenic alterations in the MMR genes MLH1, PMS2, MSH2 and MSH6, in which various types of tumors, the most prevalent being colorectal carcinoma (CRC), arise by inactivation of the second MMR allele and a subsequent high rate of repair errors during DNA replication [11]. This MMR deficiency is reflected in a high tumor mutation burden (TMB) that has been shown to benefit from immune checkpoint inhibitors [12]. The current study focuses on the clinical, histologic and molecular characterization of the G3/MMR subgroup in comparison to other glioblastoma cases from the same cohort, including cases with high TMB due to TMZ therapy. It shows the distinct characteristics of G3/MMR tumors occurring in older adults, similar to most glioblastoma cases, and reviews few additional adult cases from the literature [13,14], delineating diagnostic guidelines for the identification of these rare cases. Importantly, this is the first study showing survival advantage in adult G3/MMR glioblastoma by addition of the dual nivolumab-ipilimumab regimen of immune checkpoint inhibitors to the standard adjuvant regimen. Methods Tumor specimens, histology and immunohistochemistry (IHC). Surgical specimens were obtained from patients, as previously described [15–17]. Formalin-fixed paraffin-embedded (FFPE) sections from glioblastoma biopsies or surgical resections were stained with hematoxylin and eosin (H&E) or various antibodies for protein expression by IHC: p53 (DO-7), p16 (E6H4), Ki-67 (30-9), MLH1 (M1), PMS2 (A16-4), MSH2 (G219) and MSH6 (SP93) (Roche / Ventana Medical Systems Inc., Tucson, AZ), and GFAP (EP672Y) (Ventana / Cell Marque, Rocklin, CA), as described [5,16]. Images were acquired with Nikon Eclipse Ci microscope equipped with Nikon Digital Sight DS-Fi2 camera (Nikon Instruments Inc., Melville, NY) [4]. An integrated histologic and molecular diagnosis, according to the most recent WHO guidelines [2] was obtained for all cases, prior to classifying them in the G1–G7 molecular subgroups [4,5]. Next generation sequencing (NGS), copy number variation (CNV), and transcriptomics. The DNA and RNA NGS analyses were performed from FFPE samples by using the xT-648-gene panel (Tempus Labs, Chicago, IL), as previously described [15,17,18]. For each case, the same FFPE block was used for DNA and RNA extraction to allow direct comparison of results. Variant and CNV assessment and interpretation, and the RNA expression analysis were performed as extensively described elsewhere [15,18]. Assessment of mutation germline origin. Genetic testing from blood was performed for patient F70. For patients M62, M68 and M72, the germline origin was inferred by comparing the variant allele fraction (VAF) for the MMR mutations and the VAF for somatic heterozygous mutations (e.g. PTEN, PIK3CA, TP53), in parallel with CNV analysis. A clear distinction between the VAF in the germline versus somatic range was present in each of these cases (see Suppl. Table S1). When possible, this method was extended to the rest of MMR mutations from tumors not mapping to the G3/MMR subgroup (see Suppl. Table S2). This assessment based on VAF showed reliable somatic versus germline prediction on the NGS platform used in the cases for which genetic testing was performed in parallel [15,17–19]. Statistical analysis. Survival Kaplan-Meyer curves were analyzed by Log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests for statistical significance, as described [4,5]. Parametric unpaired t-test with Welch’s correction was performed by using GraphPad Prism (Version 10.2.3, GraphPad Software, La Jolla, CA). Statistical significance was considered for p < 0.05, and confidence intervals were 95 % for all tests. Data were analyzed and plotted by using Microsoft Excel (Microsoft Corp., Redmond, WA), and GraphPad Prism. Results Dismal prognosis of G3/MMR glioblastoma patients: treatment of one patient with nivolumab-ipilimumab immune checkpoint inhibitors improves outcome. In a prospectively assembled cohort of 218 glioblastoma patients where all the tumors were subjected to DNA and RNA NGS, and classified in molecular subgroups according to the MAPK-based G1–G7 subgroup classification [4,5], five cases with germline mutations in MMR genes stood apart as a separate subset, G3/MMR (Fig. 1A). The G3/MMR subgroup represented approximately 2 % of the glioblastoma cohort, and showed distinctive clinical, histologic and molecular characteristics, summarized in Table 1. The G3/MMR patients frequently had a clinical history of prior cancers, sometimes with multiple different malignancies, the most commonly reported being CRC in two of the five patients. Skin cancer, either melanoma or basal cell carcinoma, was also relatively common in these patients, and lymphoma and thyroid carcinoma less common (Table 1). Figure 1: Location of G3/MMR molecular subgroup tumors
A. Pie chart distribution (%) of 218 prospective glioblastoma cases in G1–G7 molecular subgroups. Purple arrow shows the G3/MMR subgroup. GBM, glioblastoma; EGFR↑, EGFR-amplified; EGFRm, EGFR-mutated. B. Regional distribution of the glioblastomas from the G3/MMR, as compared to the rest of the cohort (GBM). CC, corpus callosum. C. Selected MRI images showing multifocality (red arrows) and brainstem infiltration (blue arrow) in three G3/MMR cases.
The survival in glioblastoma depends on many factors, and these include tumor location and subsequent extent of surgical resection, age and comorbidities, accessibility to and effectiveness of adjuvant treatment [20,21]. The intrinsic aggressivity of the tumor may also influence survival [5,21]. The location of the tumors varied, with a high percentage of tumors showing midline location and multifocality, the latter often interpreted as metastatic disease (Table 1, Fig. 1B–C). The two multifocal tumors located in the midline that were only biopsied associated with the worst survival in patients M62 and M72 (Table 1, Fig. 1C). Patient M79, with peripherally located tumor crossing the midline, and receiving gross total resection and standard radiation-TMZ adjuvant therapy, marginally survived longer. Patient M68 underwent subtotal resection of the larger right frontal focus (Fig. 1C), and had slow post-surgical recovery, during which the residual tumor focus progressed. The radiation and immune checkpoint inhibitor therapy with pembrolizumab was initiated 1.5 months post-surgery and did not stop tumor growth, being aborted after 3 weeks. The median survival calculated from the date of the surgery was 3.25 months for the G3/MMR subgroup patients, and 9 months for the rest of the cohort (Table 1). The survival was equally dismal for four out of the 5 G3/MMR patients, within a range between 1.7 and 4.5 months, with only F70 surviving to 10 months. The age range between 62 and 79 years within the G3/MMR subgroup did not appear to influence the survival, as patients in their 60s or 70s performed the same in this subgroup (Table 1). However, age-adjusted survival showed a highly significant difference between patients older or younger than 50 years in the rest of the cohort, with 8 versus 20 months median survival values, respectively (Fig. 2A). The median age of the G3/MMR patients was comparable to the ≥ 50-years age-adjusted cohort, but the median survival was significantly lower, at 3.25 months versus 8 months, respectively (Fig. 2A). Compared to the three major glioblastoma subgroups, a statistically significant difference was apparent between G3/MMR patients and the G1/EGFR and G3/NF1 subgroup patients that showed 11.3- and 8.5-month median survival, respectively, but not the G7/Other subgroup patients that showed a lower median survival of 7 months (Fig. 2B). Figure 2: Survival of G3/MMR subgroup patients
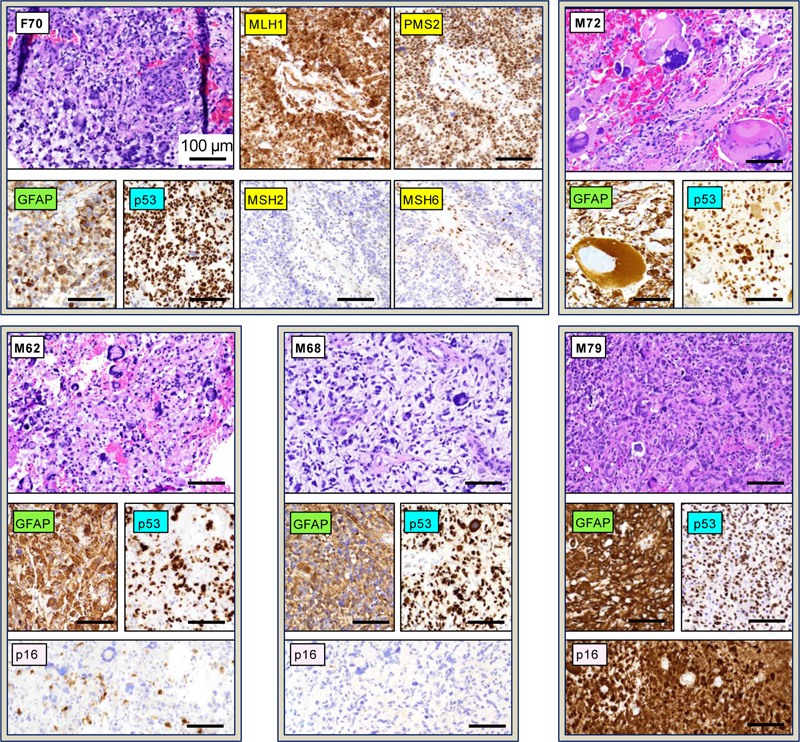
A-B. Survival curves for G3/MMR patients in comparison to patients from the rest of the cohort: older than 50 years (≥50 yo) or younger than 50 years (< 50 yo) (A), and from other G1–G7 subgroups (B). Statistical significance: *, p < 0.05; **, p < 0.01; ***, p < 0.001. N, number of deaths; n, number of patients in follow-up. The median survival in months, and the median age in years, are indicated. C. Timeline of disease progression and treatment for the G3/MMR patient F70. GBM, glioblastoma; STR, subtotal resection; ICPI, immune checkpoint inhibitors. D. Expression of the CTLA4, PD-L1, PD-1 and IDO1 immune therapy response targets in the 5 G3/MMR tumors. Levels were normalized to the lower expression values for the respective immune target. Note very high and high expression levels for IDO1 and CTLA4, respectively, in M62 and M72 tumors. F70 was the only patient from the G3/MMR subgroup with significantly longer survival to 10 months (Table 1, Fig. 2C). She had family history of first-degree cousin with glioblastoma, and history of colorectal cancer 10 years prior, status post resection and 5-flurouracil chemotherapy. She presented with dizziness, unsteady gait, sleepiness and confusion and received subtotal resection of the 7.7 cm, right temporal lobe glioblastoma, leaving residual enhancing and non-enhancing tumor in the anterior mesial temporal region. Surgery was followed by the standard radiation-TMZ regimen consisting of radiation with concurrent TMZ. A regimen of immune checkpoint inhibitors consisting of combination nivolumab-ipilimumab (Opdivo-Yervoy), in the dosing and schedule as for metastatic non-small cell lung cancer (NSCLC) [22], was then added for the following 4 months and interrupted when the patient showed decline of cognitive skills. The patient survived an additional 4 months post-therapy. The gene expression for various immune checkpoint inhibitor targets is one of the modalities used for assessing immune checkpoint inhibitor therapies [23]. The expression of CTLA4, the CD274/PD-L1 and PDCD1/PD-1 interactors, and IDO1, a negative immunomodulator in glioblastoma [24,25], showed variable levels in the G3/MMR tumors (Fig. 2D). All markers were low in the M68 tumor. IDO1 showed very high levels in the M62 and M72 tumors and low levels in the rest. The CTLA4 levels were also slightly elevated in the M62 and M72 tumors. The PD-L1 and PD-1 levels were variable, with at least one slightly elevated in the M62, M72 and M79 cases. MMR glioblastoma shows multinucleated giant cells (MGCs) and IHC compatible with p53 mutation and MMR deficiency. Histologic examination of the G3/MMR tumors showed presence of MGCs in all cases (Table 1, Fig. 3). However, except for case M72 that was diagnosed as giant cell glioblastoma, the MGCs were either scattered or focal in the other cases and admixed with neoplastic cells with high-grade neuroendocrine (HGNE) or gemistocytic morphology, which were the predominant histologic patterns. Moreover, myxoid extracellular matrix, a feature present in many tumors with HGNE morphology from the G7 / Other molecular subgroup [4], was present in three cases, M62, M68 and F70. The GFAP expression was variable, being lost in subsets of neoplastic cells, in correlation with the HGNE morphology [4]. Figure 3: Histology and IHC of G3/MMR molecular subgroup tumors
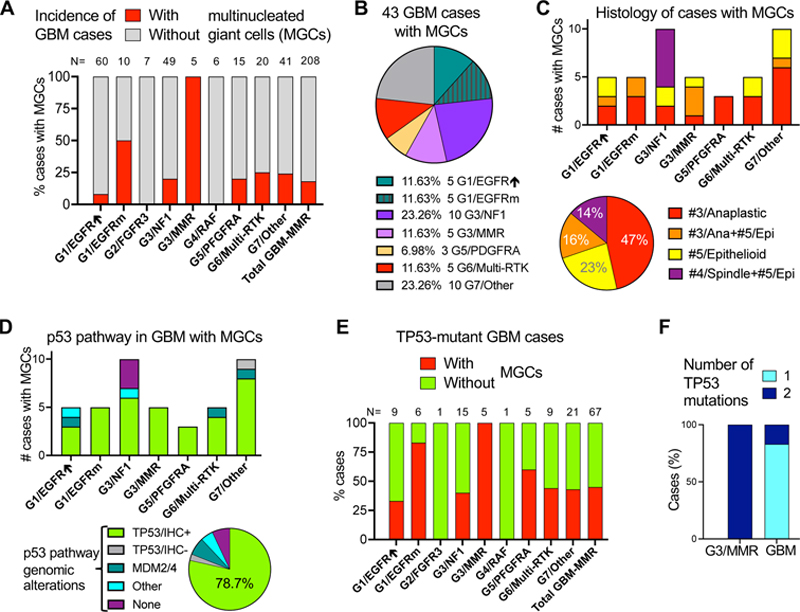
H&E panels showing tumor morphology with presence of MGCs for all 5 cases, as indicated in the upper left corner. IHC with color-coded antibodies: yellow for MMR, green for GFAP, blue for p53, and pink for p16. The MMR IHC is shown only for the F70 case and is representative for the other G3/MMR cases. All panels are shown at 200x magnification, and the scale bars are all the same, at 100 μm. Note gigantic cells in the M72 tumor, the only of the 5 G3/MMR tumors diagnosed as giant cell glioblastoma. IHC for the MMR proteins MLH1, PMS2, MSH2 and MSH6 consistently showed preserved nuclear staining for the MLH1-PMS2 pair and loss of nuclear staining for the MSH2-MSH6 pair, or just MSH6 for M68 (Table 1, Fig. 3). NGS confirmed germline MSH2 or MSH6 alterations, showing perfect correlation with MMR IHC. Importantly, the combined IHC and NGS workup revealed the MMR deficiency for the first time in 4 of 5 patients, including in patients with prior history of cancer (Table 1), underscoring the importance of combined IHC and NGS workup for glioblastoma. All the tumors showed diffuse nuclear p53 immunostaining, suggestive of TP53 mutation. The Ki-67 proliferation index was variable, showing moderate elevation of approximately 20 % in M62 and M79 tumors and high rate of approximately 50 % in the M68 and F70 tumors. IHC with p16 antibody, performed in the M62, M68 and M79 tumors, showed a different expression pattern for each case (Fig. 3). This ranged between strong expression for M79, lack of expression for M68, and mixed expression for M62, correlating with the intact, deleted, and mutant status of the CDKN2A gene, respectively (Suppl Table S1). MGC histology associates with TP53 mutation in multiple glioblastoma subgroups. The morphologic classification of glioblastoma in 12 patterns assigned to 5 histologic clusters, #1/Astrocytic (previously called EGFR-like), #2/Small neuronal, #3/Anaplastic, #4/Spindle and #5/Epithelioid, has been previously described [4]. Histologic examination of the entire cohort showed that MGCs were also present in tumors from other molecular subgroups, making up approximately one fifth (18 %) of the total glioblastoma cases other than G3/MMR ones (Fig. 4A). Tumors containing scattered MGCs resembling the G3/MMR tumors were especially enriched in the G1/EGFR-mutant subgroup (50 %), and less in the G6/Multi-RTK (25 %), G7/Other (24 %), G3/NF1 (20 %) and G5/PDGFRA (20 %) subgroups, with the major G1/EGFR-amplified subgroup showing only few cases (8 %). In total, 43 primary glioblastoma cases featured MGCs, with the G7/Other and G3/NF1 subgroups contributing 10 cases each, and the G3/MMR subgroup, G1/EGFR-amplified, G1/EGFR-mutant and G6/Multi-RTK subgroups contributing 5 cases each (Fig. 4B). These data indicated that the presence MGCs in a tumor is not specific for the G3/MMR subgroup. Figure 4: MGCs in glioblastoma: distribution in G1–G7 molecular subgroups, histologic profiles, and p53 pathway mutations
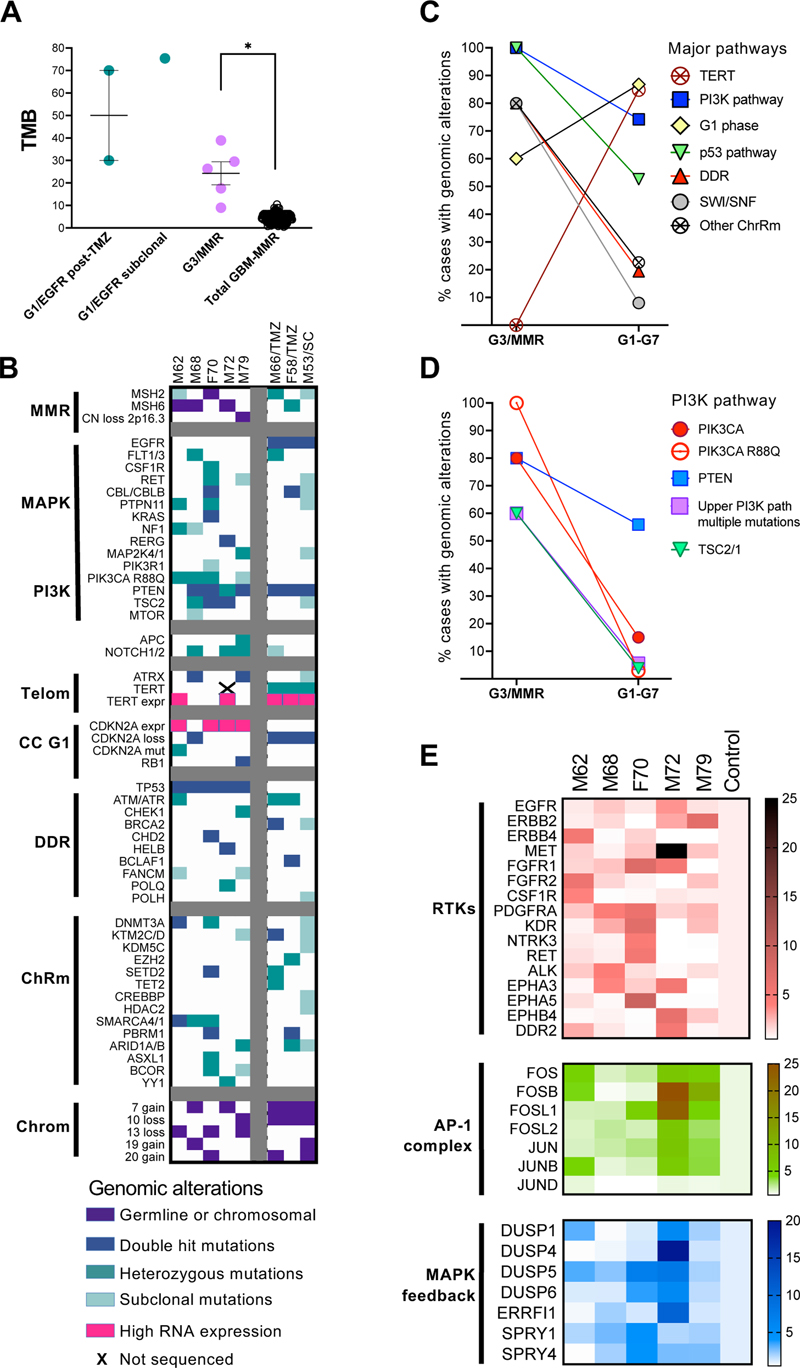
A. Incidence of cases with MGCs in the G1–G7 glioblastoma subgroups. GBM, glioblastoma; EGFR↑, EGFR-amplified; EGFRm, EGFR-mutated; Total GBM-MMR, GBM cases without the G3/MMR cases. B. Pie-chart distribution (%) of the 43 cases with MGCs in G1–G7 molecular subgroups. C. Distribution of the 43 cases with MGCs into histologic clusters in the G1–G7 molecular subgroups (upper bar graph), or as % (lower pie chart). #, number of cases; Ana, anaplastic; Epi, epithelioid. D. Distribution of p53 pathway alterations in the 43 tumors containing MGCs. TP53/IHC+ and TP53/IHC-, MGC cases with TP53 mutations and positive or negative p53 IHC, respectively; Other (than TP53 or MDM2/4) alterations include one case each with RPL5 and EBF1 mutations. E. Bar graph showing the % of cases with or without MGCs from the total GBM cases with TP53 mutations (N) and per G1–G7 molecular subgroup. Total GMB-MMR, TP53-mutant cases without the G3/MMR cases. Note that overall, only 45 % of the TP53-mutant tumors contain MGCs. F. Bar graph showing the number of allelic TP53 mutations. GBM, TP53-mutant glioblastoma cases without the G3/MMR cases. Examination of the associated morphologies in these cases revealed that they either fell into the #3/Anaplastic, #5/Epithelioid, mixed Anaplastic/Epithelioid or mixed Spindle/Epithelioid clusters, indicating that anaplastic and epithelioid morphologies are dominant in the cases with MGCs (Fig. 4C). Moreover, the mixed Spindle/Epithelioid morphology was limited to G3/NF1 subgroup tumors that may show a SEGA-like histologic profile [16]. Other than this SEGA-like morphology that was noted only in the G3/NF1 subgroup, and the giant cell glioblastoma case M72 that was unique to the cohort, the other cases showed scattered MGCs, significantly smaller than in the M72 case (Fig. 3), regardless of the molecular subgroup. No giant cells were recorded in cases from the minor subgroups G2/FGFR3 and G4/RAF, and only 5 cases mapped to the largest molecular subgroup G1/EGFR-amplified, as these three subgroups predominantly display morphologies in the #1/Astrocytic or #2/Small neuronal clusters that were virtually exclusive of MGCs. All the cases in the G3/MMR subgroup showed IHC positive for p53 (Fig. 3 and 4D), and carried TP53 mutations (Suppl. Table S1). Analysis of the other cases with MGCs showed that the large majority also showed p53 IHC positivity and carried TP53 mutations, regardless of the molecular subgroup (Fig. 4D). One tumor occurring in a patient with Li-Fraumeni syndrome showed frameshift mutation in the beginning of TP53 gene with lack of p53 protein expression, and another 12 % of cases with MGCs showed alterations in mediators of the p53 pathway, such as MDM2, MDM4, RPL5 or EBF1 [5,26,27] (Fig. 4D). Only 7 % of the cases with MGCs did not show alterations in the p53 pathway, and they all clustered in the G3/NF1 subgroup. In total, approximately one fifth (21.3 %) of the cases with MGCs were negative for p53 IHC (Fig. 4D, pie chart). Examination of the TP53 mutations in the glioblastoma cohort showed some distinctive features of the G3/MMR cases. Whereas all the cases in the G3/MMR subgroup showed TP53 mutations and MGCs, only approximately half of the cases with TP53 mutations from other molecular subgroups showed an MGC phenotype, except for the G1/EGFR-mutant subgroup that showed 83 % of TP53 mutant cases displaying an MGCs (Fig. 4E). Overall, the G3/MMR cases represented one seventh (14.3 %) of the cases showing both MGCs and p53 immunopositivity. A major difference also consisted in the number of the TP53 mutations: whereas in G3/MMR cases, TP53 was inactivated by different missense mutations most likely targeting both alleles (Suppl. Table 1), 83 % of the tumors in the rest of the cohort showed only one TP53 mutation, with the second allele inactivated by loss or neutral loss of heterozygosity (Fig. 4F). Molecular landscape of the G3/MMR glioblastoma subgroup tumors. The TMB of the G3/MMR cases was significantly higher than that of the other glioblastoma cases, with or without high MSI (Table 1 and Fig. 5A). For comparison, three additional cases from the glioblastoma cohort showed very high TMB (Fig. 5A; Suppl. Table S2). One was a tumor at initial presentation in a 53-years-old male (M53/SC) that showed clonal VAF in few mutations common to G1/EGFR-amplified tumors, and subclonal VAF in a multitude of less common mutations indicative of a subclonal population of cells with very high TMB. Two other cases were recurrent, G1/EGFR-amplified subgroup tumors in male and female patients aged 66 and 58 years, respectively (M66/TMZ and F58/TMZ), previously treated with TMZ, a known hypermutator phenotype inducer [28]. Interestingly, of the 20 post-TMZ, recurrent tumors subjected to NGS in this cohort, of which only 5 mapped to the G1/EGFR-amplified subgroup, both tumors with hypermutator phenotype belonged to the G1/EGFR-amplified subgroup. All these three G1/EGFR-amplified tumors harbored very high TMB, stable MSI, but lacked MGC histology. Their immunohistochemical profiles were undistinguishable from those of G1/EGFR-amplified tumors, showing negative p53 and p16 IHC. Figure 5: Molecular profiling of the G3/MMR subgroup
A. TMB mean±SEM graph of individual case values from indicated cases. Post-TMZ, post TMZ chemotherapy; Total GBM-MMR, GBM cases without the G3/MMR cases. Statistical significance: *, p < 0.05. B. Genomic alterations in effectors of the pathways indicated on the left are shown by color-coded squares. Telom, telomere maintenance; CC G1, cell cycle G1 phase; DDR, DNA damage response; ChRm, chromatin remodeling; Chrom, chromosomes; expr, RNA expression. C–D. Line graphs tracing the incidence of the indicated alterations in the G3/MMR subgroup in comparison with the rest of the G1–G7 molecular subgroups. E. RNA expression heatmaps for the indicated RTKs, transcription factor AP-1 complex and MAPK pathway feedback effectors. A comparison of the molecular landscape of the G3/MMR cases with the 3 cases with very high TMB and, in general, with the distribution of mutations from the rest of the G1–G7 molecular subgroups (Suppl. Tables S2-S3), showed distinctive features for the G3/MMR subgroup (Table 1; Fig. 5B–D; Suppl. Table S1). Germline pathogenic alterations in MSH2 or MSH6 MMR genes were present in all G3/MMR cases but absent in the rest of the cohort. These were either splice site or frameshift mutations, resulting in truncation of the protein, or loss of the MSH2/MSH6 locus on chromosome 2p16.3, an exceedingly rare event in glioblastoma present only in the M79 G3/MMR case from the entire cohort. Interestingly, the MSH6 p.F1088fs mutation was recurrent, either as germline or as second hit somatic mutation (Suppl. Table S1). In comparison, the 3 cases with very high TMB showed pathogenic or likely pathogenic MSH2 or MSH6 mutations with VAF in the somatic range, of which only one resulted in protein truncation, in M66/TMZ, the other being missense mutations, with subclonal VAF in the M53/SC tumor (Suppl. Table S2). In comparison, a paucity of pathogenic mutations in MMR, POLE and MUTYH genes, the latter two also known to induce hypermutator phenotype [29,30], was apparent in the cohort, without high TMB in any of these mutant cases (Suppl. Table S2). In particular, the G2/FGFR3 molecular subgroup displayed higher incidence of MMR/MUTYH mutations, with the M60 case showing the only MSH6 truncating mutation with VAF in the somatic range from the cohort. As previously described, this tumor showed a typical G2/FGFR3 molecular profile and lacked MGCs [15] (Suppl. Table S2). A truncating mutation in MLH3, a paralog of MLH1, with VAF in the germline range, in the G1/EGFR-amplified M31 tumor, was the only other germline MMR truncating mutation from the cohort, and did not show high TMB, most likely due to compensation by the MLH1-PMS2 MMR pair. The only two tumors that showed concomitant MGCs, TP53 mutations and mutations in either MUTYH or MSH3 but without hypermutator phenotype belonged to the G5/PDGFRA subgroup (Suppl. Table S2). It has been shown that deficiency in DNA repair results in a specific DNA mutation signature in tumors, represented by predominance of C:G to T:A transitions in MMR-deficient tumors, and C:G to A:T transversions in POLE, POLD1 and MUTYH mutant tumors [31,32]. All G3/MMR tumors, as well as the M66/TMZ and F58/TMZ tumors, showed an MMR-type DNA mutation signature (Table 1). Surprisingly, the M53/SC tumor showed a POLE-type DNA mutation signature, and mutation analysis revealed two subclonal POLH mutations, including one pathogenic (Fig. 5B, Suppl. Table S2). Since no report is available in the literature for association of POLH mutations with high TMB, the latter may be due to the MSH2 subclonal mutations but possibly fine-tuned by the POLH deficiency. The G3/MMR subgroup tumors lacked or showed low incidence for many of the common glioblastoma molecular alterations, such as simultaneous chromosome 7 gain and 10 loss (see also [5]), TERT promoter mutations and CDKN2A homozygous loss (Fig. 5B–C, Suppl. Table S3). Conversely, they showed 100 % TP53 mutation rate and high incidence of mutations in genes from DNA damage response (DDR) and chromatin remodeling pathways, especially from the SWI/SNF complex (Fig 5B–C, Suppl. Tables S1, S3). Interestingly, likely pathogenic NOTCH1 missense mutations were detected in three of the five G3/MMR cases. 100 % of G3/MMR TP53 mutations were biallelic (Fig. 4F) and targeted Arg residues (Suppl. Table S3). In the rest of the cohort, TP53 Arg mutations were seen in a quarter of cases, the highest rate of 40 % being noted in the G7 / Other subgroup. The PI3K pathway was strongly activated by mutations in PIK3CA and PTEN upstream effectors, but also by TSC2 mutations (Fig. 5B–D, Suppl. Table S1). The 80 % mutation rates for PIK3CA and PTEN were the highest in the cohort, the PIK3CA mutation rate being three-fold higher than the next highest rate from the G5/PDGFRA subgroup (Suppl. Table S3). Three G3/MMR cases showed overlapping PIK3CA and PTEN mutations, and one of these showed additional PIK3R1 subclonal mutation (Fig. 5B). The G3/MMR 60 % rate of overlapping mutations in PI3K pathway upstream effectors was 10-fold higher than the 5.6 % rate in the rest of the cohort (Fig. 5D). Strikingly, PIK3CA p.R88Q was the activating mutation in all four G3/MMR mutant cases, whereas it was a single event in the 34 PIK3CA-mutant cases from the rest of the cohort (Fig. 5B, D; Suppl. Table S3). Similar to TP53, PIK3CA mutations on Arg residues were scarce in the rest of the cohort, numbering one p.R88Q and one p.R38L in the G7 / Other and G3/NF1 subgroups, respectively. PTEN mutations were biallelic in two G3/MMR cases (40 %), whereas two concomitant PTEN mutations were noted only in 3 cases in the rest of the cohort (2.5 %), all showing subclonal VAF, suggestive of heterogeneity rather than of biallelic hit. Double hit mutations were also detected in TSC2 tumor suppressor gene, in two out of three mutant G3/MMR cases (Fig. 5B, Suppl. Table S1). TSC1/2 mutations are relatively uncommon in glioblastoma, numbering 8 cases in the rest of the cohort (3.8 %), without double hit (Fig. 5D). The MAPK pathways was activated by mutations in many effectors, some overlapping, such as PTPN11 and NF1 or KRAS (Fig. 5B). None of the major RTKs defining the G1/EGFR, G2/FGFR3, G5/PDGFRA or G6/Multi-RTK subgroups was amplified or mutated. Likely pathogenic missense mutations in RET in two cases, and FLT1 and CSF1R in one case each, were detected (Suppl. Table S1), of which the RET mutation in case F70 was accompanied by overexpression (Fig. 5E, Suppl. Table S4). Virtually all the G3/MMR cases showed moderate overexpression of at least one RTK except for EGFR, and the M72 tumor showed high MET overexpression close to the range of the MET-amplified tumors from G6/Multi-RTK subgroup (Fig. 5E, Suppl. Table S4). FOS and JUN families of transcription factors heterodimerize to form the activator protein-1 (AP-1) complex that is phosphorylated and activated by MAPK [33]. The four members of the FOS family are transcription activators, whereas JUND from the JUN family acts mostly as repressor. The expression of the AP-1 complex transcription factors in G3/MMR cases showed overexpression of one or more activating transcription factors, with the M72 and M79 tumors highly overexpressing FOSB, and additionally FOSL1 for M72 (Fig. 5E, Suppl. Table S5). Consistent with its role as suppressor, JUND showed no overexpression and was decreased in the M68 and F70 tumors that showed lesser overexpression of the transcription activator family members (Suppl. Table S5). The activation of the MAPK and PI3K canonical growth pathways has been shown to result in an inhibitory feedback response in glioblastoma [15]. The dual-specificity phosphatases (DUSP) directly dephosphorylate and inactivate MAPK, with DUSP5 and DUSP1/6 specifically dephosphorylating ERK1 and ERK2, respectively, and DUSP4 dephosphorylating ERK1, ERK2 and JUNK [34]. Of these, DUSP5 was upregulated in all five G3/MMR tumors, and DUSP1 in three tumors (Fig. 5E and Suppl. Table S6), suggesting MAPK activation in all tumors. Moreover, the more specific inhibitors ERRFI1 and SPRY1/4 targeting downregulation of growth signaling from the EGFR and FGFR RTK families, respectively [35,36], were strongly upregulated in M72 and F70 tumors, respectively (Fig. 5E and Suppl. Table S6). Discussion The identification of molecular subgroups susceptible to targeted therapies is the ultimate goal of tumor classifications. The MAPK pathway-based G1–G7 molecular classification of glioblastoma allowed subclassification of the small number of cases with genetic MMR deficiency in the G3/MMR molecular subgroup [5]. The incidence of these tumors in the prospective cohort of 218 glioblastoma cases is low, approximately 2 %. Unlike three recent studies that have characterized few glioblastoma cases with MMR deficiency mainly in younger adults [13,14,37], this is the first study assembling five cases of glioblastoma with germline MMR deficiency in adults over 60 years of age, who represent the large majority of patients with glioblastoma. Of the studies in younger MMR patients, the only one showing survival data concluded that these MMR patients exhibit better survival by comparing them to a standard glioblastoma cohort significantly much older, with a difference of 13 years in median age [13]. Studies, including the current one, have clearly shown significant age-dependent differences for glioblastoma patient survival [20,21], and therefore the conclusion of a longer survival even for younger adult MMR patients is inaccurate methodologically in the afore mentioned study [13]. The current study unequivocally showed dismal survival for the adult G3/MMR glioblastoma patients, significantly shorter than for the rest of the glioblastoma cohort adjusted for age. This finding stratifies the G3/MMR as a high-risk glioblastoma subgroup and aligns it with a recent study reporting poor prognosis for Lynch-syndrome patients with IDH-mutant astrocytoma [38]. The speedy identification of the G3/MMR patients is peremptory for prompt therapy with immune checkpoint inhibitors. The only controlled study to date of immune checkpoint therapy for recurrent MMR-deficient glioblastoma has not found a significant difference to control when using pembrolizumab, a PD-1 inhibitor [39]. In addition, a recent case report has shown lack of effectiveness of pembrolizumab for controlling Lynch-syndrome-associated glioblastoma in a young adult patient [40]. In agreement with these data, the M68 patient from the G3/MMR subgroup was briefly treated with pembrolizumab without response, although it is not clear if the lack of immune checkpoint marker expression may have contributed to the inhibitor resistance. In contrast, the F70 patient who was treated with the dual regimen of nivolumab-ipilimumab after standard concurrent radiation-TMZ adjuvant therapy, showed much better survival than the other patients from the G3/MMR subgroup. This result, even if unique to this adult G3/MMR patient, shows that the combination nivolumab-ipilimumab targeting both PD-1 and CTLA-4, respectively, may be effective on adult G3/MMR glioblastoma. Few pediatric examples of MMR/Lynch syndrome glioblastoma were successfully treated with PD-1 inhibitors [41,42], but in one case, a sustained response could only be achieved after addition of anti-CTLA-4 ipilimumab to the initial anti-PD-1 nivolumab regimen [43]. This dual regimen is also used in metastatic CRC associated to Lynch syndrome [44], and as first-line therapy for metastatic or advanced NSCLC with TMB ≥ 10 mutations / MB, regardless of the PD-L1 expression levels [22,45,46]. The identification of the G3/MMR patients relies on clinical, histologic and molecular clues. A review of the literature identified 5 additional adult (≥ 50-years) G3/MMR cases with clinical, histologic and molecular data [13,14], and the integrated findings from the total of 10 G3/MMR adult glioblastoma patients is summarized in Table 2. Clinical history of prior cancers pertains to half of the patients, with highest frequency of CRC, in 4 of the 10 cases. Older G3/MMR patients appear to be prone to multifocal or deep-seated tumors that warrant subtotal resections or only biopsies. Histologically, 100 % of these tumors contain MGCs. As shown in this study, MGCs are not specific for G3/MMR tumors, and other G1–G7 subgroups, especially the ones with tumors displaying morphology classified in the anaplastic and epithelioid clusters, may show a variable number of tumors with MGCs (see Fig. 4). In addition to MGCs, all the G3/MMR tumors also show positive p53 IHC. However, as shown in this study, nearly 80 % of the tumors with MGCs showed p53 immunopositivity, with the G1/EGFR-mutant molecular subgroup mimicking the closest the G3/MMR subgroup in terms of morphology and p53 immunopositivity (see Fig. 4). Overall, the G3/MMR subgroup cases represented 14.3 % (one seventh) of the total glioblastoma cases showing both MGCs and p53 immunopositivity. The IHC with the MMR panel appears to be specific for G3/MMR cases (Table 2), but will only be yielding positive results in 1 out of 7 glioblastoma cases with concomitant MGCs and p53 immunopositivity.
The NGS molecular analysis with TMB inclusion is necessary for detecting and/or confirming the presence of G3/MMR cases. In general, high TMB is a relatively rare occurrence in untreated, first-presentation glioblastoma, and is more often seen in recurrences with TMZ-related hypermutator phenotype [28]. Noteworthy, both post-TMZ recurrences described here mapped to the G1/EGFR-amplified subgroup, suggesting an association worth looking into. A hypermutator phenotype in untreated tumors may be the result of germline and/or somatic mutations in MMR, POLE or MUTYH genes. Germline MMR mutations were found only in MSH2 or MSH6, with the MSH6 p.F1088fs mutation recurrent in 2 studies, the current one and that by Kim et al. [14]. In the current study, somatic MMR mutations did not result in hypermutator phenotype in untreated, first-presentation glioblastomas (see Suppl. Table S2). However, at least three ≥ 50-year-old patients in the studies by Hadad et al. and Kim et al. presented with hypermutator phenotype in sporadic glioblastomas with somatic MMR alterations [13,14]. The question arises if these tumors should be included in the G3/MMR high-risk subgroup where MMR mutations are causative, or if their tumors, like the M53/SC case, show only added hypermutator phenotype to a baseline mutation core that fits another G1–G7 subgroup and show therefore better prognosis. More studies are necessary to clarify this issue, and treatment with immune checkpoint inhibitors may prove beneficial in glioblastomas with both germline and somatic MMR mutations. For MUTYH, a pan-cancer recent study has shown that biallelic mutations and not heterozygous germline variants result in high TMB [30]. Glioblastoma cases with high TMB due to MUTYH mutations have not been reported in the literature, and in our series, the two tumors with heterozygous germline MUTYH mutations did not show high TMB (see Suppl. Table S2). POLE mutations in glioblastoma are equally rare, with only one in this cohort, without high TMB. However, POLE somatic mutations with hypermutator phenotype and usually accompanied by germline or somatic MMR mutations have been reported mainly in young glioblastoma patients [13,29]. Beside high TMB, the G3/MMR subgroup tumors showed a common signature represented by mutations resulting in RAS activation, with high incidence of PTPN11 and NF1 mutations (Table 2), an association also noted in the G3/NF1 subgroup [4]. The resulting MAPK activation was accompanied by high levels of the transcription factors from the AP-1 effector complex and by upregulation of a negative feedback, of which DUSP5 was the most consistently upregulated. The activation of the PI3K pathway was present in all the cases, with high incidence of PIK3CA mutations, especially of p.R88Q, of overlapping PIK3CA and PTEN mutations, and of TSC2 mutations (Table 2 and Fig. 5D). As shown, TP53 was mutant in all the cases, with predominance of biallelic Arg mutations. Very high incidence of mutations in the DDR and chromatin remodeling pathways, particularly in DNMT3A, SETD2 and the SWI/SNF complex, coupled to absence of the characteristic TERT promoter mutations or concomitant chromosome 7 gain and 10 loss, distinguished the G3/MMR subgroup from the other G1–G7 subgroups (Table 2 and Fig. 5C) [5]. Mutations in APC and NOTCH1/2 genes were also overrepresented in G3/MMR patients from this study and that by Kim et al. [14]. In general, the G3/MMR molecular profile resembled closer the profiles of other solid cancers and was highly divergent from that of non-MMR glioblastoma [47]. Conclusions In conclusion, this study characterizes rare cases of adult glioblastoma associated to Lynch syndrome that appear to represent a high-risk G3/MMR subgroup showing distinctive clinical, histologic and molecular characteristics. It reviews the available cases in the literature, and proposes immune checkpoint inhibitors as adjuvant treatment, particularly the dual nivolumab-ipilimumab regimen that showed net survival benefit in the G3/MMR patient receiving it. More studies are necessary to validate this regimen and correlate it with the molecular parameters usually used in CRC associated to Lynch syndrome, such as TMB, MSI status and immune checkpoint markers expression levels. Ethics, Consent and Permissions This study was conducted in accordance to the Declaration of Helsinki, and approved by the Ethics Committee of NeuroMarkers (Protocol code NM-REC-2 on 9 January 2020 and 30 January 2023) for studies involving human subjects. Informed consent was obtained for all subjects. Consent for Publication Patient consent for publication was waived due to the study not identifying the patients. Availability of Data and Materials The genomic and transcriptomic datasets supporting the conclusions of this article are de-posited in the public repository NeuroMarkers Open Research Database https://neuromarkers.org/database under the accession numbers G-NM/24-1 and T-NM/24-1, respectively. Conflicts of Interest Statement The author declares no conflicts of interest. Funding Statement This work was supported by an award from NeuroMarkers [NM2024-1] to M.-M.G. Acknowledgements This paper is dedicated to the patients and their families. References 1. Ostrom, Q.T.; Truitt, G.; Gittleman, H.; Brat, D.J.; Kruchko, C.; Wilson, R.; Barnholtz-Sloan, J.S. Relative survival after diagnosis with a primary brain or other central nervous system tumor in the National Program of Cancer Registries, 2004 to 2014. Neurooncol Pract 2020, 7, 306-312. https://doi.org/10.1093/nop/npz059 2. WHO Classification of Tumors Editorial Board. Central Nervous System Tumors, 5th ed., International Agency for Research on Cancer, Lyon (France), 2021. https://doi.org/10.1093/neuonc/noab106 3. Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes & Cancer 2010, 1, 1170-1177. https://doi.org/10.1177/1947601911407325 4. Georgescu, M.M. Multi-Platform Classification of IDH-Wild-Type Glioblastoma Based on ERK/MAPK Pathway: Diagnostic, Prognostic and Therapeutic Implications. Cancers (Basel) 2021, 13. https://doi.org/10.3390/cancers13184532 5. Georgescu, M.M. Translation into Clinical Practice of the G1–G7 Molecular Subgroup Classification of Glioblastoma: Comprehensive Demographic and Molecular Pathway Profiling. Cancers (Basel) 2024, 16. https://doi.org/10.3390/cancers16020361 6. Li, Y.M.; Suki, D.; Hess, K.; Sawaya, R. The influence of maximum safe resection of glioblastoma on survival in 1229 patients: Can we do better than gross-total resection? J Neurosurg 2016, 124, 977-988. https://doi.org/10.3171/2015.5.JNS142087 7. Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005, 352, 987-996. https://doi.org/10.1056/NEJMoa043330 8. Oster, C.; Schmidt, T.; Agkatsev, S.; Lazaridis, L.; Kleinschnitz, C.; Sure, U.; Scheffler, B.; Kebir, S.; Glas, M. Are we providing best-available care to newly diagnosed glioblastoma patients? Systematic review of phase III trials in newly diagnosed glioblastoma 2005-2022. Neurooncol Adv 2023, 5, vdad105. https://doi.org/10.1093/noajnl/vdad105 9. Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers (Basel) 2021, 13. https://doi.org/10.3390/cancers13081795 10. Dunn, D.B. Larotrectinib and Entrectinib: TRK Inhibitors for the Treatment of Pediatric and Adult Patients With NTRK Gene Fusion. J Adv Pract Oncol 2020, 11, 418-423. https://doi.org/10.6004/jadpro.2020.11.4.9 11. Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer 2015, 15, 181-194. https://doi.org/10.1038/nrc3878 12. Therkildsen, C.; Jensen, L.H.; Rasmussen, M.; Bernstein, I. An Update on Immune Checkpoint Therapy for the Treatment of Lynch Syndrome. Clin Exp Gastroenterol 2021, 14, 181-197. https://doi.org/10.2147/CEG.S278054 13. Hadad, S.; Gupta, R.; Oberheim Bush, N.A.; Taylor, J.W.; Villanueva-Meyer, J.E.; Young, J.S.; Wu, J.; Ravindranathan, A.; Zhang, Y.; Warrier, G., et al. "De novo replication repair deficient glioblastoma, IDH-wildtype" is a distinct glioblastoma subtype in adults that may benefit from immune checkpoint blockade. Acta Neuropathol 2023, 147, 3. https://doi.org/10.1093/neuonc/noad179.0670 14. Kim, H.; Lim, K.Y.; Park, J.W.; Kang, J.; Won, J.K.; Lee, K.; Shim, Y.; Park, C.K.; Kim, S.K.; Choi, S.H., et al. Sporadic and Lynch syndrome-associated mismatch repair-deficient brain tumors. Lab Invest 2022, 102, 160-171. https://doi.org/10.1038/s41374-021-00694-3 15. Georgescu, M.M.; Islam, M.Z.; Li, Y.; Traylor, J.; Nanda, A. Novel targetable FGFR2 and FGFR3 alterations in glioblastoma associate with aggressive phenotype and distinct gene expression programs. Acta Neuropathol Commun 2021, 9, 69. https://doi.org/10.1186/s40478-021-01170-1 16. Georgescu, M.M.; Li, Y.; Islam, M.Z.; Notarianni, C.; Sun, H.; Olar, A.; Fuller, G.N. Mutations of the MAPK/TSC/mTOR pathway characterize periventricular glioblastoma with epithelioid SEGA-like morphology-morphological and therapeutic implications. Oncotarget 2019, 10, 4038-4052. https://doi.org/10.18632/oncotarget.27005 17. Georgescu, M.M.; Olar, A. Genetic and histologic spatiotemporal evolution of recurrent, multifocal, multicentric and metastatic glioblastoma. Acta Neuropathol Commun 2020, 8, 10. https://doi.org/10.1186/s40478-020-0889-x 18. Georgescu, M.M.; Whipple, S.G.; Notarianni, C.M. Novel neoplasms associated with syndromic pediatric medulloblastoma: integrated pathway delineation for personalized therapy. Cell Commun Signal 2022, 20, 123. https://doi.org/10.1186/s12964-022-00930-3 19. Georgescu, M.M.; Islam, M.Z.; Li, Y.; Circu, M.L.; Traylor, J.; Notarianni, C.M.; Kline, C.N.; Burns, D.K. Global activation of oncogenic pathways underlies therapy resistance in diffuse midline glioma. Acta Neuropathol Commun 2020, 8, 111. https://doi.org/10.1186/s40478-020-00992-9 20. Alexopoulos, G.; Zhang, J.; Karampelas, I.; Patel, M.; Kemp, J.; Coppens, J.; Mattei, T.A.; Mercier, P. Long-Term Time Series Forecasting and Updates on Survival Analysis of Glioblastoma Multiforme: A 1975-2018 Population-Based Study. Neuroepidemiology 2022, 56, 75-89. https://doi.org/10.1159/000522611 21. Guo, X.; Gu, L.; Li, Y.; Zheng, Z.; Chen, W.; Wang, Y.; Wang, Y.; Xing, H.; Shi, Y.; Liu, D., et al. Histological and molecular glioblastoma, IDH-wildtype: a real-world landscape using the 2021 WHO classification of central nervous system tumors. Front Oncol 2023, 13, 1200815. https://doi.org/10.3389/fonc.2023.1200815 22. Reck, M.; Borghaei, H.; O'Byrne, K.J. Nivolumab plus ipilimumab in non-small-cell lung cancer. Future Oncol 2019, 15, 2287-2302. https://doi.org/10.2217/fon-2019-0031 23. Donisi, C.; Pretta, A.; Pusceddu, V.; Ziranu, P.; Lai, E.; Puzzoni, M.; Mariani, S.; Massa, E.; Madeddu, C.; Scartozzi, M. Immunotherapy and Cancer: The Multi-Omics Perspective. Int J Mol Sci 2024, 25. https://doi.org/10.3390/ijms25063563 24. Ozawa, Y.; Yamamuro, S.; Sano, E.; Tatsuoka, J.; Hanashima, Y.; Yoshimura, S.; Sumi, K.; Hara, H.; Nakayama, T.; Suzuki, Y., et al. Indoleamine 2,3-dioxygenase 1 is highly expressed in glioma stem cells. Biochem Biophys Res Commun 2020, 524, 723-729. https://doi.org/10.1016/j.bbrc.2020.01.148 25. Xing, Z.; Li, X.; He, Z.N.T.; Fang, X.; Liang, H.; Kuang, C.; Li, A.; Yang, Q. IDO1 Inhibitor RY103 Suppresses Trp-GCN2-Mediated Angiogenesis and Counters Immunosuppression in Glioblastoma. Pharmaceutics 2024, 16. https://doi.org/10.3390/pharmaceutics16070870 26. Dobbelstein, M.; Levine, A.J. Mdm2: Open questions. Cancer Sci 2020, 111, 2203-2211. https://doi.org/10.1111/cas.14433 27. Shen, A.; Chen, Y.; Liu, L.; Huang, Y.; Chen, H.; Qi, F.; Lin, J.; Shen, Z.; Wu, X.; Wu, M., et al. EBF1-Mediated Upregulation of Ribosome Assembly Factor PNO1 Contributes to Cancer Progression by Negatively Regulating the p53 Signaling Pathway. Cancer Res 2019, 79, 2257-2270. https://doi.org/10.1158/0008-5472.CAN-18-3238 28. Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H., et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006, 66, 3987-3991. https://doi.org/10.1158/0008-5472.CAN-06-0127 29. Erson-Omay, E.Z.; Caglayan, A.O.; Schultz, N.; Weinhold, N.; Omay, S.B.; Ozduman, K.; Koksal, Y.; Li, J.; Serin Harmanci, A.; Clark, V., et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol 2015, 17, 1356-1364. https://doi.org/10.1093/neuonc/nov027 30. Paller, C.J.; Tukachinsky, H.; Maertens, A.; Decker, B.; Sampson, J.R.; Cheadle, J.P.; Antonarakis, E.S. Pan-Cancer Interrogation of MUTYH Variants Reveals Biallelic Inactivation and Defective Base Excision Repair Across a Spectrum of Solid Tumors. JCO Precis Oncol 2024, 8, e2300251. https://doi.org/10.1200/PO.23.00251 31. Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev 2014, 24, 107-113. https://doi.org/10.1016/j.gde.2013.12.005 32. Mark, S.C.; Sandercock, L.E.; Luchman, H.A.; Baross, A.; Edelmann, W.; Jirik, F.R. Elevated mutant frequencies and predominance of G:C to A:T transition mutations in Msh6(-/-) small intestinal epithelium. Oncogene 2002, 21, 7126-7130. https://doi.org/10.1038/sj.onc.1205861 33. Song, D.; Lian, Y.; Zhang, L. The potential of activator protein 1 (AP-1) in cancer targeted therapy. Front Immunol 2023, 14, 1224892. https://doi.org/10.3389/fimmu.2023.1224892 34. Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J 2013, 280, 489-504. https://doi.org/10.1111/j.1742-4658.2012.08716.x 35. Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015, 4, 215-266. https://doi.org/10.1002/wdev.176 36. Segatto, O.; Anastasi, S.; Alema, S. Regulation of epidermal growth factor receptor signalling by inducible feedback inhibitors. J Cell Sci 2011, 124, 1785-1793. https://doi.org/10.1242/jcs.083303 37. Barresi, V.; Simbolo, M.; Mafficini, A.; Piredda, M.L.; Caffo, M.; Cardali, S.M.; Germano, A.; Cingarlini, S.; Ghimenton, C.; Scarpa, A. Ultra-Mutation in IDH Wild-Type Glioblastomas of Patients Younger than 55 Years is Associated with Defective Mismatch Repair, Microsatellite Instability, and Giant Cell Enrichment. Cancers (Basel) 2019, 11,https://doi.org/10.3390/cancers11091279 38. Suwala, A.K.; Stichel, D.; Schrimpf, D.; Kloor, M.; Wefers, A.K.; Reinhardt, A.; Maas, S.L.N.; Kratz, C.P.; Schweizer, L.; Hasselblatt, M., et al. Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 2021, 141, 85-100. https://doi.org/10.1007/s00401-020-02243-6 39. Lombardi, G.; Barresi, V.; Indraccolo, S.; Simbolo, M.; Fassan, M.; Mandruzzato, S.; Simonelli, M.; Caccese, M.; Pizzi, M.; Fassina, A., et al. Pembrolizumab Activity in Recurrent High-Grade Gliomas with Partial or Complete Loss of Mismatch Repair Protein Expression: A Monocentric, Observational and Prospective Pilot Study. Cancers (Basel) 2020, 12. https://doi.org/10.3390/cancers12082283 40. Nakase, K.; Matsuda, R.; Sasaki, S.; Nakagawa, I. Lynch Syndrome-Associated Glioblastoma Treated With Concomitant Chemoradiotherapy and Immune Checkpoint Inhibitors: Case Report and Review of Literature. Brain Tumor Res Treat 2024, 12, 70-74. https://doi.org/10.14791/btrt.2023.0042 41. Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V., et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol 2016, 34, 2206-2211. https://doi.org/10.1200/JCO.2016.66.6552 42. Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S., et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409-413. https://doi.org/10.1126/science.aan6733 43. Larouche, V.; Atkinson, J.; Albrecht, S.; Laframboise, R.; Jabado, N.; Tabori, U.; Bouffet, E.; international b, M.c. Sustained complete response of recurrent glioblastoma to combined checkpoint inhibition in a young patient with constitutional mismatch repair deficiency. Pediatr Blood Cancer 2018, 65, e27389. https://doi.org/10.1002/pbc.27389 44. Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A., et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018, 36, 773-779. https://doi.org/10.1200/JCO.2017.76.9901 45. Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E., et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2019, 381, 2020-2031. https://doi.org/10.1056/NEJMoa1910231 46. Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M., et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J Clin Oncol 2019, 37, 992-1000. https://doi.org/10.1200/JCO.18.01042 47. Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M., et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017, 23, 703-713. https://doi.org/10.1038/nm.4333
Copyright: © 2024 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |