|
|
||||||||||||||||||||||||||||||
|
Free Neuropathology 6:11 (2025) |
||||||||||||||||||||||||||||||
|
Review |
||||||||||||||||||||||||||||||
|
Selective cellular and regional vulnerability in frontotemporal lobar degeneration: a scoping review |
||||||||||||||||||||||||||||||
|
Kashif Ravasia1 and Veronica Hirsch-Reinshagen2 |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
Corresponding author: |
||||||||||||||||||||||||||||||
|
Submitted: 15 August 2024 |
||||||||||||||||||||||||||||||
|
Keywords: FTLD, Selective vulnerability, Human post mortem, Histology, Tau, TDP-43, FUS |
||||||||||||||||||||||||||||||
|
Abstract The three main types of frontotemporal lobar degeneration (FTLD) are characterized by the accumulation of abnormal proteins, namely tau, TDP-43 and FUS. The distribution of these proteins within different human brain regions is well known, as is the range of morphological variability of the cellular inclusions they form. Compared to the extensive knowledge that exists about distinct protein aggregates in FTLD, surprisingly little is known about the specific cell (sub)types that these inclusions affect. Even less is known about disease-specific abnormalities other than protein inclusions in affected and unaffected areas. These are non-trivial knowledge gaps. First, knowing which cell subtypes are vulnerable or resilient to the development of pathological protein inclusions is crucial to understand the cellular disease mechanisms. Second, mounting evidence suggests that non-cell autonomous mechanisms may play important roles in neurodegenerative conditions. For example, astrocytic tau pathology is associated with synaptic loss in corticobasal degeneration but not in progressive supranuclear palsy. Furthermore, changes that are more difficult and time-consuming to quantify, for example loss of a specific neuronal subtype that does not develop pathological inclusions, remain virtually unexplored and their relevance for disease progression are unknown. This scoping review is an attempt to collate all histological evidence from human studies that address the question of cell-specific vulnerability in the most common FTLD subtypes. By taking a systematic approach including various brain cell types such as neurons and their subtypes as well as astrocytes, microglia and oligodendrocytes and the entire central nervous system with its affected and unaffected regions, this review summarizes the current status in the field and highlights important knowledge gaps. |
||||||||||||||||||||||||||||||
|
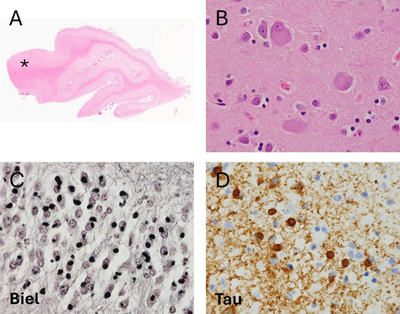
Introduction Frontotemporal dementia (FTD) is a clinical neurodegenerative syndrome characterized by prominent deficits in behaviour, language, and/or personality, with relative sparing of memory early in the disease course. FTD is the third most common form of neurodegenerative dementia after Alzheimer’s disease (AD) and dementia with Lewy bodies, and affects about 20,000 to 30,000 people in the United States according to estimates by Knopman and Roberts1 alone. This prevalence is similar to that identified in the United Kingdom2. FTD is phenotypically heterogeneous, including cases of behavioural variant FTD (bvFTD) and primary progressive aphasia (PPA), the latter of which can be additionally subclassified into nonfluent variant PPA (nfvPPA), semantic variant PPA (svPPA), or logopenic variant PPA (lvPPA)3. These subtypes can often be difficult to disentangle clinically, as additional symptoms may develop as the disease progresses, and symptoms can mimic or overlap with other neurodegenerative conditions such as AD, Parkinson’s disease (PD), or amyotrophic lateral sclerosis (ALS)3. The neuropathological condition underlying most cases of FTD, termed frontotemporal lobar degeneration (FTLD), is also highly heterogeneous. The clinical diagnosis of FTD and the neuropathological diagnosis of FTLD are not always concordant. Patients with pathological FTLD and significant loss of memory may be diagnosed in life with AD, and those with significant motor symptoms may be diagnosed with ALS or PD4,5. Conversely, some cases of clinically diagnosed bvFTD and lvPPA meet neuropathological criteria for AD6,7. As the name implies, FTLD is characterized by preferential atrophy of the frontal and temporal lobes and abnormal protein inclusions in neurons and glial cells. Historically, the first inclusions to be recognized were argyrophilic Pick bodies in Pick’s disease (PiD)8. Until the 1980s, cases with clinical FTD and a FTLD pattern of brain atrophy but without Pick bodies were described as atypical PiD cases8,9. With the advent and widespread adoption of immunohistochemistry (IHC), it became possible to identify novel protein inclusions in FTLD cases. The first one to be identified was hyperphosphorylated tau10,11, which ultimately led to a revised and expanded categorization of tau-positive FTLD, including PiD. In 2006, another protein, transactive response DNA binding protein 43 (TDP-43), was identified in about 90 % of cases of tau-negative FTLD12,13. Most of the remaining cases were later found to contain a protein known as RNA-binding protein fused in sarcoma (FUS)14,15. While there remains a small group of cases that stain positive for ubiquitin but negative for tau, TDP-43, and FUS, over 99 % of FTLD cases can now be classified into tauopathies (FTLD-tau), FTLD with TDP-43 pathology (FTLD-TDP), and FTLD with FUS pathology (FTLD-FUS)15. Each of these neuropathological entities exhibits known patterns of neuronal and glial inclusions16–18 in the neocortex, deep grey nuclei and infratentorial structures. In the different FTLD pathologies, the selective vulnerability of different brain cell types such as neuronal subtypes, astrocytes, microglia and oligodendrocytes is incompletely understood. Selective neuronal vulnerability has been a focus of recent research on neurodegenerative diseases including AD and PD. In each of these syndromes, specific sets of neurons degenerate more quickly and more consistently than other neuronal populations19,20. Furthermore, in each disease affected neuronal populations display similar alterations in organelle distribution, neurotransmitter receptors, electrophysiology, and/or morphology19,20. This patterned neurodegeneration has been less studied in FTLD. In addition, FTLD pathology prominently affects glial cells whereas in AD or PD the pathological inclusions are mainly neuronal. The pathophysiological effects of the glial involvement in FTLD are largely unknown. Pathological glial involvement has been shown in amyotrophic lateral sclerosis (ALS), where progressive motor neuron degeneration has been shown to be modulated by non-neuronal cells through a process known as non-cell autonomous neurodegeneration21,22. Therefore, studies that seek to understand the processes responsible for patterned neurodegeneration should consider and include the evaluation of non-neuronal dysregulation as one possible pathological mechanism of disease progression. Here, we review the available literature on selective neuronal vulnerability in FTLD and include data on glial pathology and its relationship to neuronal pathology wherever possible. The review is organized primarily according to the type of FTLD proteinopathy and the FTD disease subtype. This review places special emphasis on human post mortem studies involving colocalization of pathology using IHC or immunofluorescence, as these studies allow identification of specific cell populations and definitive neuropathological diagnosis of each case. In the IHC studies described below, the most commonly used subtype-specific markers include parvalbumin and calbindin for inhibitory neurons and subtype-specific neurotransmitters or enzymes involved in neurotransmitter metabolism such as choline acetyltransferase (ChAT) or tyrosine hydroxylase (TH). Glial cell-specific markers include glial fibrillary acidic protein (GFAP) and aquaporin 4 (AQP4) as pan-astrocytic markers, and vimentin and CD44 as markers of activated or reactive astrocytes. Microglial markers include cluster of differentiation 68 (CD68), human leukocyte antigen – DR isotype (HLA-DR) and ionized calcium-binding adapter molecule 1 (Iba1). 1. Frontotemporal lobar degeneration with tau pathology Tauopathies, named for the accumulation of microtubule-associated protein tau, account for approximately 40 percent of FTLD cases23. While numerous tauopathies have been described and characterized, this review focuses on the three FTLD tauopathies for which the existing literature is most robust: Pick’s disease (PiD), corticobasal degeneration (CBD), and progressive supranuclear palsy (PSP). 1.1 Pick’s Disease 1.1.1 General features PiD typically presents with bvFTD or lvPPA, and the gross appearance involves frontotemporal atrophy with relative sparing of the Rolandic cortex and the posterior superior temporal gyrus24 (Figure 1). Neuropathologically, PiD demonstrates astrocytosis, loss of neurons, ballooned neurons with eosinophilic cytoplasm, and extensive spongiosis25 (Figure 1). Most strikingly andof pathognomic value, PiD demonstrates Pick bodies, i.e. round, solitary, argyrophilic neuronal cytoplasmic inclusions (NCIs) primarily located in pyramidal neurons and in the dentate granular neurons of the hippocampus26 (Figure 1). Glial pathology has also been observed in PiD cases , including ramified astrocytes and small oligodendroglial inclusions24. Pick bodies consistently display 3-repeat (3R) tau pathology, while astrocytes display a variable combination of 4R and 3Rtau16,25,27,28, depending on the cases studied. Many of the studies evaluating the selective cellular vulnerability in PiD were conducted before the modern classification scheme for FTLD making it difficult to confirm whether these cases actually correspond to PiD. Figure 1. Pathological features of Pick’s Disease.
A. Low power image reveals relative preservation of the posterior aspect of the superior temporal gyrus (asterisk, 50x). B. Hematoxylin and eosin stain reveals neocortical ballooned neurons (600x). C. Bielschowsky silver stain reveal numerous argyrophilic round inclusions (Pick bodies) in the granular neuronal cells of the hippocampal dentate fascia (600x). D. Tau immunohistochemistry reveals numerous tau-immunoreactive Pick bodies and granular neuropil staining (600x). 1.1.2 Neuronal pathology Table 1 provides a summary of the known neuronal subtypes that bear tau pathology and/or undergo selective neurodegeneration in PiD. Many regions of the frontal and temporal cortex display marked neuronal loss which begins in the outer cortical layers and later progresses to involve the deeper layers of the cortex and extracortical regions25. Pyramidal cell density is most markedly reduced in lamina II of the cortex, with lesser involvement of laminae III and V, and occurs in the early stages of the disease29. This finding is consistent with studies of laminar distribution of tau pathology in PiD, which have demonstrated that Pick bodies are prominent in laminae II and III, with variable involvement of deeper cortical layers25,30.
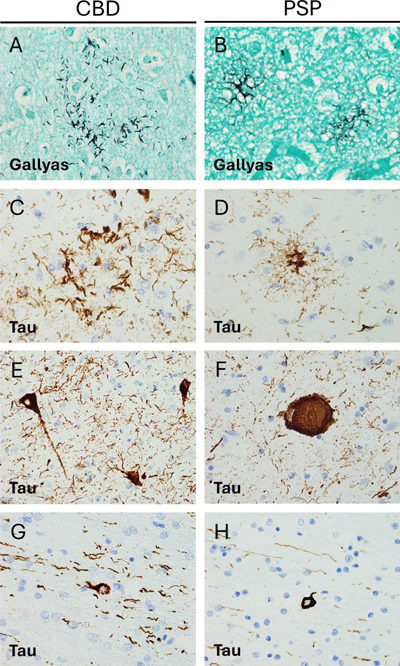
Von Economo neurons (VENs) are found in lamina V of the anterior cingulate and frontoinsular regions of the cortex and are morphologically distinct from neighboring pyramidal neurons31. VENs have been shown to be significantly affected in confirmed PiD cases and degenerated more rapidly than neighboring cells32. Moreover, VENs demonstrate tau aggregation and cytoplasmic swelling in all stages of PiD31, which is consistent with the observation that early neuron loss and astrocytosis are most significant in the orbitofrontal and mediofrontal cortices in this disease25,33. In the hippocampus, the dentate gyrus is most significantly involved in PiD34, with greater tau pathology in granule than in hilar cells35. One study found a trend toward decreased calbindin immunoreactivity in dentate granule cells in PiD, as compared with other pathological subtypes of FTLD. This trend did not reach statistical significance, and loss of immunoreactivity may also be secondary to generalized neuronal atrophy rather than to selective vulnerability of dentate cells36. As early as 1998, it was observed that the striatum can exhibit atrophy in severe or longstanding cases of PiD, with greater involvement of the caudate than the putamen26. These findings have been confirmed in a more recent study on the progression of PiD, in which the researchers also detected late developing tau depositions in the globus pallidus25. These tau inclusions are primarily intra-neuronal, but the phenotypic features of the affected neurons have not been studied in detail25. Limited evidence suggests the mediodorsal nucleus of the thalamus is preferentially affected in PiD37. Furthermore, asymmetric thalamic atrophy has been observed by volume-based magnetic resonance imaging (MRI)38, but the affected neuronal populations remain unknown. The brainstem can also be neuropathologically affected in PiD. In one study, pontine involvement was found to develop after the onset of limbic and neocortical disease, and the serotonergic and noradrenergic nuclei of the raphe nuclei and locus coeruleus exhibited more severe pathology relative to the remaining brainstem25. Involvement of the substantia nigra, the dorsal motor nucleus of the vagus nerve and other brainstem nuclei was observed to coincide with pontine pathology, while involvement of the inferior olivary nuclei and medullary pyramids occurred only in advanced disease stages25. There is rare literature on the involvement of the cerebellum in PiD. In 1999, Braak et al. determined that mossy fibers, monodendritic brush cells, and dentate projection neurons were involved in a set of cases classified as PiD, but these findings have not been replicated yet39. In summary, the distribution of tau pathology and neuronal loss in the cortex of patients with PiD seem to be correlated, particularly with regard to VENs. Yet, the relationship between tau pathology and rate of loss of different neuronal populations is still unknown for the remaining brain areas such as hippocampus, brainstem and cerebellum, where the distribution of the PiD tau pathology has been described in detail. 1.1.3 Glial involvement PiD cases display significant astrogliosis and astroglial tau pathology in cortical regions. Astroglial tau pathology is most notable in the orbitofrontal cortex and mediofrontal cortices, which show significant neuronal loss and tau-immunopositive ramified astrocytes even in early stages of disease25. In early stages of PiD, IHC for GFAP, a marker that is increased in reactive astrocytes, demonstrates widespread astrocytic reaction in laminae I, III, and IV33. Some of these astrocytes also stain positive for tau, with one double-labelling IHC experiment showing that 23 % of cortical GFAP-positive astrocytes were also positive for tau and that tau-positive filamentous inclusions can sometimes displace GFAP-positive fibrils29. Interestingly, markers of astrocytic apoptosis and dysregulated ceramide metabolism, thought to be neuroinflammatory and pro-apoptotic, have been found in regions of neuronal loss in PiD33,40. Oligodendroglial coiled bodies are restricted to areas affected by neuronal tau pathology and degeneration in PiD41. Interestingly, a PiD-specific oligodendroglial inclusion has been described24,41, but its relationship to oligodendrocyte degeneration or axonal loss is unknown. Similar to astrogliosis, microgliosis has been consistently described in the frontal and temporal cortex of PiD for decades. This finding has been confirmed in cases meeting revised criteria following the development of the modern FTLD classification system13,18,42. Cortical microglial cells demonstrate activation by enhanced staining for HLA-DR29 and this increased microglial activation is especially notable in areas displaying a high burden of Pick bodies43. The extent of grey matter microglial involvement in PiD seems to be quite substantial. Indeed, comparative studies have shown a significative increase in grey matter microgliosis not only relative to controls but also relative to cases of CBD, PSP, and rare tauopathies42. These differences are observed regardless of the IHC protocol used to identify microglia, including CD68, Iba1, and CR3/43, a novel antibody that reacts with the human leukocyte antigen isotypes DR, DP, and DQ42. Additional studies consistently show microgliosis in the white matter underlying the frontal and temporal cortex29,42,43. When activated microglia are measured specifically, whether by using CD68 or HLA-DR, more gliosis is seen in the white matter than in the grey matter29,42. By contrast when total microglial burden is measured using Iba1, the difference between these regions is less evident42. Microglial dystrophy is also more severe in subcortical white matter than in the cortical grey matter, with many microglia demonstrating a loss of fine branches and unusual cytoplasmic morphology42. Together, these data suggest that neuronal loss, astroglial reactivity and tau pathology significantly yet not perfectly overlap in the cortex in PiD. How these three alterations relate to each other has not been described in detail. Furthermore, microglial activation and dystrophy seem greater in the white matter than in the overlying cortex. The relationship between microglial alterations, oligodendroglial changes, axonal and neuronal loss as well as tau pathology remains unclear. Finally, despite the early and consistent involvement of the hippocampus, and the known involvement of subcortical structures later in PiD, there are no additional details on glial involvement and their relationship to neuronal tau pathology in these areas. 1.2 Corticobasal Degeneration 1.2.1 General features Corticobasal degeneration (CBD) is a 4R tauopathy initially described as the underlying pathological condition of a clinical syndrome now termed corticobasal syndrome (CBS). CBS is characterized by asymmetric rigidity and apraxia, dystonia, myoclonus, and cortical symptoms44. However, it is now understood that CBD can also present clinically as nfvPPA, progressive supranuclear palsy syndrome (PSPS), frontal behavioural-spatial syndrome (FBS), or the classic CBS motor syndrome45–47. Moreover, cases of clinically diagnosed CBS have been shown on post-mortem pathological examination to meet diagnostic criteria for PSP, PiD, AD, Creutzfeldt-Jacob disease, and FTLD-TDP48,49. Neuropathologically, CBD is characterized by cortical atrophy that tends to involve the peri- Rolandic cortex but can also involve temporal regions associated with language, and anterior frontal regions associated with behaviour and personality50. CBS cases exhibit cortical spongiosis and astrogliosis of the superficial laminae and ballooned neurons in laminae III, V, and VI. Diagnostic tau-immunoreactive pathology includes neurofibrillary tangles, numerous thread-like lesions in white and grey matter50 and characteristic glial lesions such as astrocytic plaques and oligodendroglial coiled bodies51 (Figure 2). Figure 2. Pathological features of Corticobasal degeneration and Progressive supranuclear palsy.
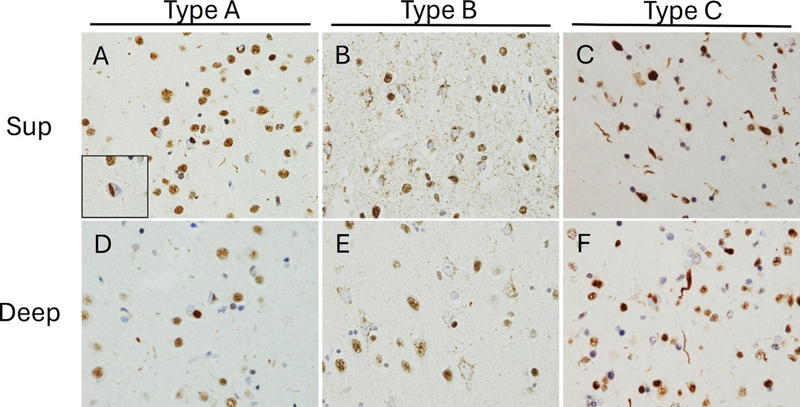
A, B. Gallyas staining reveals different argyrophilic astrocytic inclusions. In CBD (A) only the distal processes of astrocytes are stained, whereas in PSP (B) tufted astrocytes display increased proximal cytoplasmic staining (600x). C, D. Tau immunohistochemistry (IHC) reveals differences in astrocytic morphology between CBD and PSP with astrocytic plaques in the former (C) and tufted astrocytes in the latter (D) (600x). E, F. Tau IHC highlights neurofibrillary tangle pathology in both conditions, with globose tangles (F) being more frequent in PSP. Note increased background thread pathology in CBD (C, E) (600x). G, H. Oligodendroglial coiled bodies are seen on tau IHC in both conditions (600x). 1.2.2 Neuronal pathology Table 1 provides a summary of the known neuronal subtypes that bear tau pathology and/or undergo selective neurodegeneration in CBD. CBD is characterized by atrophy of the frontal, parietal and temporal cortex, with consistent involvement of the premotor cortex52. In keeping with the diverse range of clinical presentations, 4R-tau can accumulate in a variety of neuroanatomical regions. In classical CBS cases, 4R-tau shows peri-Rolandic distribution46, but there are also cases with predominant temporal involvement46,53 and some with an unusual degree of frontal tau pathology53. Cortical neurons demonstrate ballooning and achromasia in deep cortical layers and a variety of tau immunoreactive pathology ranging from granular pre-tangles to more filamentous neurofibrillary tangles51,54. Small neurons in upper cortical layers are most vulnerable to CBD51. In some cases of CBD, hippocampal neurons in the CA2 region and the dentate gyrus can show tau pathology 50. The extent of tau pathology varies by clinical presentation, being more pronounced in PSPS than in CBS despite pathological confirmation of CBD in both sets of cases46. CBD also exhibits extensive pathology in the basal ganglia. Filamentous neuronal inclusions are often visible in the caudate and the putamen, but less consistently seen in the globus pallidus51. Neuronal pathology tends to become more severe with disease progression49,52. Confirmed cases of CBD often exhibit mild-to-moderate neuronal loss and tau-positive neuronal inclusions and threads in the thalamus55, which is consistently affected in its ventrolateral portion56. In CBD, the brainstem is not classically considered as a region of interest, but the substantia nigra can be affected. In these cases, the substantia nigra appears markedly depigmented with pale intracytoplasmic inclusions in surviving neurons57, numerous pre-tangles, and severe loss of dopaminergic and GABAergic neurons but without significant astrocytic plaque pathology51,58,59. Some CBD cases also show tau deposition in the tegmentum and inferior olivary nucleus. This phenomenon has not been studied in detail, but current evidence suggests that medullary tau deposition is more common in cases that clinically present as PSPS46. The cerebellum has not been extensively studied in CBD, although variable neuronal loss and gliosis were found in the cerebellar dentate nucleus together with scattered cortical Purkinje cell axonal torpedoes and mild Bergmann gliosis51. One study found the cerebellum to be involved in approximately half of the cases studied60. Cerebellar involvement mainly consisted in diffuse granular accumulation of cytoplasmic tau in the cell bodies of Purkinje cells, and of doughnut-shaped structures in the cerebellar molecular layer in a smaller set of cases60. While the latter alterations were not studied directly in any CBD case, ancillary studies in PSP cases revealed their location in the GFAP-positive radial processes of Bergman’s glia60. A relatively detailed map of neuronal tau-positive pathology has been described in CBD, but the degree of correlation between tau-positive pathology and stereotactically measured neuronal loss remains uncertain, especially in subcortical and infratentorial regions. It also remains unclear whether tau-positive pathology predominates in specific subtypes of affected neurons. It would also be of particular interest to understand these relationships in early and later disease stages. 1.2.3 Glial involvement Glial pathology in CBD is of diagnostic significance51. Indeed, the characteristic thread pathology of CBD is likely predominantly glial rather than neuronal, as only a small fraction of thread-like structures are double labeled with neurofilament antibodies41. Moreover, studies of astrocytic tau pathology provide evidence of glial cell involvement in regions that are preferentially affected by CBD. Astrocytic plaques have been shown to co-localize with CD44, possibly suggesting a reactive change61, but not with GFAP62. Furthermore, the presence of astrocytic plaques in specific areas correlates with neuronal loss and reduced local density of HOMER1+ excitatory post-synaptic puncta, providing evidence for a relationship between glial and neuronal pathology63. One study has described tau-positive astrocytic plaques in the superior frontal gyrus prior to the development of symptomatic neurodegeneration64, and another study demonstrated that astrocytes and neurons in the grey matter of the anterior frontal lobe demonstrate tau pathology even in preclinical CBD65. A semiquantitative score for astrocytic plaque density was shown to remain moderate throughout disease progression, while the density of neuronal inclusions in the anterior frontal grey matter increased with disease progression65. Taken together, these findings have led some scholars to speculate that CBD is a primary astrogliopathy65,66. Astrogliosis and astrocytic plaques have also been observed in the hippocampus67, and are consistently found in the basal ganglia of confirmed CBD cases. Astrocytic plaques can be found throughout the striatum, although they are more numerous in the caudate than the putamen65. Indeed, the caudate is even more severely affected than the anterior frontal gyrus in preclinical CBD65. It is also worth noting that these astrocytic plaques develop early in the disease process, and one study has found that basal ganglia astrocytic pathology is most severe in the preclinical stage of CBD, with diminished density of plaques in end-stage disease65. These findings suggest an early involvement of basal ganglia astrocytes, in keeping with the frequently observed clinical picture49. Astrocytic plaques have also been described in the thalamus, although they are less frequent than in the neocortex or the caudate58. Finally, GFAP-positive radial processes of Bergman’s glia60 may show doughnut-shaped tau-positive structures in the cerebellar molecular layer in a small set of CBD cases60. Oligodendroglial coiled bodies are distributed extensively throughout affected areas in CBD, but they are less frequent than in PSP41. Furthermore, little is known about the relationship between these oligodendroglial coiled bodies and other histological aspects such as myelin density or axonal density in the surrounding area68. Microglial activation is observed in confirmed CBD cases when assessed by CD68 immunoreactivity42. In one large comparative study of frontotemporal microglial burden, CD68-positive microglia were significantly more numerous in the frontal grey matter than in the temporal grey matter of CBD cases. There was however no significant increase in the density of CR3/43- or Iba-1-immunoreactive microglia42. Additionally, the parietal somatosensory and the superior temporal cortex demonstrate more widespread microgliosis in CBD than in PSP56. White matter microgliosis is a consistent finding in CBD56. Significant differences between controls and CBD cases have been observed in subcortical white matter in the frontal, temporal, and parietal lobes42,56. The frontal and temporal subcortical regions display moderate-to-severe microglial dystrophy that is more noticeable in the white matter than in the associated cortical grey matter42. Activated microglia are also widely distributed throughout the basal ganglia, with HLA-DR immunostaining demonstrating their significant proliferation in the striatum, the lentiform nucleus, the subthalamic nucleus, and the substantia nigra, as compared to controls56. Microglial activation is also observed in the ventrolateral portion of the thalamus56. The early and region-specific presence of astrocytic pathology as well as the link between astrocytic and synaptic pathology raises the possibility that astrocytic tau pathology may be pathogenic in CBD. It is therefore somewhat surprising that no more detailed studies exist correlating subtype-specific neuronal loss with astroglial tau pathology. Notably, neuronal tau pathology has been described in areas without significant astrocytic pathology such as the brainstem, suggesting that different pathomechanisms may be at play in these regions. Microglial reactivity seems to mirror neuronal pathology in CBD. It endeavors to further dissect the relationship between astrocytic and microglial pathology to understand whether microglial activation reflects a primary neuroinflammatory mechanism or a specific response to neuronal injury. 1.3 Progressive Supranuclear Palsy 1.3.1 General features PSP is also a 4R tauopathy and presents most often as a movement disorder, yet cognitive decline is quite common and can be the presenting feature45. While the classic PSPS involves vertical gaze palsy, unprovoked falls, akinesia, and cognitive dysfunction, each of these symptoms can present along a spectrum of severity, and some cases with predominant akinesia or cognitive involvement are clinically diagnosed as CBS or PPA69,70. Neuropathologically, PSP is characterized by globose neurofibrillary tangles, thin, branching astrocytic tau inclusions (“tufted astrocytes”), and oligodendroglial coiled bodies (Figure 2). The basal ganglia and brainstem tend to be especially involved, though cases with features suggestive of frontotemporal dementia often demonstrate substantial cortical involvement8. 1.3.2 Neuronal pathology Table 1 provides a summary of the known neuronal subtypes that bear tau pathology and/or undergo selective neurodegeneration in PSP. Historically, PSP was thought to be primarily a disease of the basal ganglia and the midbrain with cortical involvement being limited and largely confined to the pre-central gyrus71. More recent studies have however challenged this view. Indeed, many PSP cases demonstrate widespread frontal and temporal atrophy, and these cases often manifest clinically with cognitive, behavioural, and linguistic symptoms similar to those observed in other forms of FTLD72,73. Immunohistochemical studies have demonstrated tau-positive neuronal tangles and neuropil threads in the superior frontal gyrus, middle frontal gyrus, and inferior temporal gyrus, with greater cortical tau pathology in cases that clinically manifest with frontotemporal dementia as compared to classical PSPS74. Analyses using confocal microscopy have shown that both excitatory and inhibitory cortical synapses are reduced in pathologically confirmed cases of PSP with frontal tau pathology63. In contrast to CBD, astrocytic pathology in PSP does not appear to correlate locally with loss of synapses63, suggesting a possible divergence in the mechanisms of synaptic vulnerability between the two diseases. While many of the classical motor deficits in PSP are related to subcortical pathology, the primary motor cortex and supplementary motor areas are often affected in PSP as well. Corticocortical projection neurons in the pre-supplementary motor area and inhibitory interneurons in the primary motor cortex have been identified as particularly vulnerable populations in PSP75, but pyramidal neurons also display variable degrees of pathology76. Compared to cases presenting clinically with CBS or PPA, cases presenting with classical PSPS have been found to exhibit greater pyramidal motor neuron involvement76. Hippocampal involvement in PSP remains poorly characterized in the literature. Preliminary studies have demonstrated enlarged neurons and neurofibrillary tangles in the parahippocampal gyrus (PHG) and in the CA1 sector of the hippocampus77. Neuronal tau pathology appears to precede astroglial or oligodendroglial involvement in the hippocampus, and the burden of neuronal pathology can be quite severe78. By contrast, basal ganglia have been studied extensively in PSP with consistent findings of early neuronal and glial tau pathology throughout the striatum, globus pallidus, and subthalamic nucleus8. GABA is the primary neurotransmitter involved in basal ganglia circuitry and decreased expression of GAD-67, a marker of GABAergic interneurons, has been confirmed in case-control studies79. However, the relationship between neurofibrillary tau pathology and affected neuronal subtypes has not been evaluated. Additionally, the nucleus basalis of Meynert exhibits mild-to-moderate neuronal loss and a reduction in ChAT positivity has been shown in at least some cases. Altogether however, the basal forebrain is only modestly affected in PSP in comparison to other neurodegenerative conditions80. PSP cases demonstrate both neuronal loss and microglial activation in many regions of the thalamus. In particular, the intralaminar nuclei appear to be profoundly affected, with one case-control study reporting a loss of 45 % of neuronal density across the centromedian and parafascicular nuclei in PSP cases81. The ventral lateral nucleus also exhibits atrophy and neuron loss, particularly in cases with greater involvement of the primary motor cortex75. The brainstem exhibits striking changes in PSP. Typically, both divisions of the substantia nigra are affected. In the pars reticularis (SNr), there is a loss of overall neuron density and decreased parvalbumin reactivity among surviving neurons, suggesting particularly pronounced vulnerability among the parvalbumin-positive cells82. This selective involvement of circuits involving parvalbumin-positive neurons has also been observed in Parkinson’s disease (PD). Yet, PSP cases appear to exhibit more severe disruptions to the parvalbumin-positive interneurons and more frank atrophy than PD cases82. In the pars compacta (SNc), there is a duration-dependant, selective dropout of neuromelanin-positive cells82. Indeed, for reasons that are poorly understood, dopaminergic cells appear profoundly vulnerable to the changes induced by PSP. Tyrosine hydroxylase-immunoreactive cells (TH-IR) in the nearby A10 region including the midline ventral tegmental area and the parabrachial pigmented nucleus are also affected with the loss of approximately 50 % of TH-IR neurons compared with controls83. Disruptions to dopaminergic signalling have been experimentally linked to downregulation of parvalbumin circuitry in mouse models, providing a potential explanation for the selective vulnerability of these two distinct neuronal populations84. PSP cases also often demonstrate marked but selective neuronal loss in the locus coeruleus and the mesencephalic motor nuclei85,86. The locus coeruleus displays marked loss of noradrenergic neuromelanin-positive neurons explaining the relative pallor visible on gross inspection. Quantification using IHC has revealed a loss of 49 % of neuromelanin-positive neurons relative to controls86. Cholinergic neurons in the mesopontine nuclei, including the lateral dorsal tegmental nucleus and the pedunculopontine nucleus (PPN) are also affected80. More recently, these results have been replicated in a case-control study conducted by Sébille et. al. (2019), showing that PSP cases exhibit greater neuronal loss than controls in both the PPN and the cuneiform nucleus85. Both cholinergic neurons, identified using IHC for ChAT, and non-cholinergic neurons are affected, and the PPN is more severely affected in PSP than in PD85. Notably, the study found minimal neuronal loss in the surrounding regions85, supporting the hypothesis that disease propagation is not driven by anatomical proximity alone. There is evidence to suggest moderate involvement of the cerebellum in most clinical phenotypes of PSP78. There is also increasing awareness of a rare clinical presentation of PSP with predominant cerebellar ataxia, which tends to exhibit more pronounced cerebellar neuron loss, tau-positive granular profiles in Purkinje cells, and grumose degeneration in the dentate nucleus87. Neuronal involvement has been evaluated in greater detail in PSP than in CBD and PiD. It is interesting to note that inhibitory neurons seem to be affected at least in the cortex, basal ganglia, and substantia nigra in PSP. In addition, other neuronal subtypes such as cholinergic and dopaminergic also seem affected, suggesting that neurotransmitter subtype is not the defining feature of vulnerable neurons to PSP pathology. Based on this, transcriptomic studies may provide additional insights into the similarities of vulnerable neuronal subpopulations in PSP. 1.3.3 Glial involvement Astrocytic pathology in PSP is complex and intriguing, as there is not always a direct relationship between astrogliosis and the presence of tufted astrocytes. For example, one study showed that despite a very substantial burden of tau-immunoreactive tufted astrocytes in the motor cortex, many cases exhibit only minimal gliosis when evaluated with GFAP88. This finding cannot be attributed to variation between cases, as the cases demonstrated remarkable homogeneity in the pattern of gliosis. Tufted astrocytes, similar to astrocytic plaques, have been shown to co-localize with CD4462, possibly suggesting a reactive change61, but not with GFAP. Some research has been conducted into the involvement of astrocytes and oligodendrocytes in the subcortical white matter, and no significant difference has been demonstrated in the burden of GFAP or myelin basic protein (MBP) between PSP cases and controls89. Curiously, one biochemical study found that insoluble tau was detectable in white matter regions by Western blotting despite the absence of tau immunostaining in contiguous sections90. Finally, evaluation of oligodendrocyte-specific pathology suggests that PSP is not a primary oligodendrogliopathy, in contrast to multiple system atrophy and globular glial tauopathy91. Astrocytic pathology in the basal ganglia appears to be an early event, with particularly severe early astrocytic involvement in the striatum78,92. The discrepancy between GFAP distribution and astroglial tau pathology has also been described in the basal ganglia. In one study, the caudate and putamen exhibited the highest burden of tufted astrocytes, while the globus pallidus and substantia nigra exhibited most astrogliosis88. No relationship has been found between astrogliosis or neuronal tau pathology and tufted astrocyte density. Yet, the severity of astrogliosis has been shown to correlate with the density of neurofibrillary tangles88,93. Characteristic astroglial and oligodendroglial inclusions, i.e. tufted astrocytes and coiled bodies, respectively, are consistently found in the thalamus of moderate-to-severe PSP cases. Conditional probability analyses suggest that in most cases thalamic glial inclusions occur later than striatal inclusions but earlier than neocortical inclusions78. Analysis of astrocytic pathology has revealed both astrogliosis and tufted astrocytes in midbrain regions, including the tectum and the red nucleus88. Astroglial pathology appears more limited in the pons and medulla, with mild astrogliosis and very few tufted astrocytes78,88. Microglial activation has also been demonstrated in PSP cases relative to controls. In PSP, the frontal cortex exhibits statistically significant microgliosis that can be detected using immunostaining for HLA-DR or Iba-142,56. The microgliosis is often most severe in the motor cortex56, and microglial pathology in this region correlates with neuronal pathology in the same region56. Interestingly, the somatosensory cortex also exhibits statistically significant microgliosis56. While involvement of the neocortex in PSP is not as pronounced as in CBD42,56, the presence of microglial activation suggests that it may be worthwhile to more thoroughly investigate cortical pathology in PSP. PSP cases often demonstrate white matter microgliosis, but the extent and distribution of the latter vary widely across studies. Evidence of increased overall microglial density is conflicting, with one study finding an increase in Iba-1 positive microglia in the frontal white matter and another study finding no increase in Iba-1 positive cells despite an increase in CD68 positivity42,89. Evidence of activation is also conflicting, as some studies but not other ones have demonstrated significantly increased microglial burden in the frontal and temporal white matter when assessed with immunostaining for CD68 and HLA-DR and compared with controls42,43,56. In the basal ganglia, microgliosis can be extensive, with significant elevations in HLA-DR-positive microglial burden throughout the globus pallidus and subthalamic nucleus56. Microglial activation in the thalamus also appears to be widespread when assessed using HLA-DR immunostaining, with increased burden in the ventrolateral nucleus and anterior nucleus when compared to controls or CBD cases. Adjacent structures, such as the mammillothalamic tract and the thalamic fasciculus, also demonstrate increased microglial burden56. Finally, the brainstem shows robust microglial activation in the superior colliculus, the medial longitudinal fasciculus, the substantia nigra, the red nucleus, and the pontine base as measured by HLA-DR expression56. Overall, there is robust knowledge on the distribution of astroglial and microglial activation in PSP. Less understood is the relationship between astrocytic tau pathology and astrocyte reactivity and how these latter relate to microgliosis. There seems to be at least some correlation between neuronal tau pathology and microgliosis. The relative independence of neuronal tau pathology and microgliosis from astrocytic pathology, as exemplified in the brainstem, is intriguing and requires further exploration. In addition, detailed glial transcriptomic phenotyping may help to identify the astrocytic populations responsible for regional astrogliosis and those most vulnerable to accumulation of 4R-tau. Yet, it is also possible that astrocytes undergo proteomic changes and loss of GFAP positivity as tau accumulates. This highlights the need for additional studies evaluating glial involvement in PSP and its relationship to neurodegeneration. 2. Frontotemporal lobar degeneration with TDP-43 pathology TDP-43 was identified in 2006 as the pathological protein present in most cases of ubiquitin-positive, tau-negative FTLD12,13. As a result of this discovery, cases that had previously been described as FTLD-U (for ubiquitin) were reclassified as FTLD with TDP-43-immunoreactive pathology (FTLD-TDP). A harmonized histologic classification system for FTLD-TDP now exists with four well-defined subtypes lettered A-D and a more recently discovered, rapidly progressive phenotype provisionally labelled “type E”94,95. FTLD-TDP type D is very rare and only found in familial cases with a mutation in the valosin-containing protein (VCP) gene94. FTLD-TDP type E is also rare and considered by some authors as a variant of type B96. The present review will focus on types A, B, and C, as they collectively account for the significant majority of FTLD-TDP cases8,94 (Figure 3). Figure 3. TDP-43 immunohistochemical features of FTLD-TDP subtypes.
A,D. FTLD-TDP type A shows compact neuronal cytoplasmic inclusions (NCIs) and short neurites that predominate in superficial over deep neocortical layers. Neuronal intranuclear inclusions (inset in A) are a distinguishing feature (600x). B, E. Type B pathology shows frequent granular NCIs that affect all neocortical layers (600x). C, F. Type C pathology is identified by long, frequently corkscrew-like, neuritic inclusions that preferentially affect superficial cortical layers, but can also affect the deep cortex (600x). Sup = superficial. 2.1 FTLD-TDP type A 2.1.1 General features Clinically, FTLD-TDP type A often presents as bvFTD or nvPPA, though there can also be motor neuron involvement8. An heritable form of FTLD-TDP type A caused by a mutation in the progranulin gene (GRN) on chromosome 17, and the familial variant often demonstrates more widespread pathology and a younger age at death97,98. Moreover, another subset of FTLD-TDP cases is associated with a pathological C9orf72 hexanucleotide repeat expansion on chromosome 9. Most of these cases fulfill histological criteria of type B, or an overlap between type A and type B (type AB). Only a minority present histologically as pure type A99. FTLD-TDP type A is characterized by moderate or numerous compact neuronal cytoplasmic inclusions (NCIs) and dystrophic neurites in layer II of the neocortex94 (Figure 3). Lentiform neuronal intranuclear inclusions (NIIs) are also commonly identified, but they are usually much less numerous than NCIs100. Late-stage disease often results in neuronal death, loss of positivity for neuronal IHC markers such as NeuN, and, importantly, clearance of inclusions101. This can complicate research findings, as TDP-43 positivity can be lost in end-stage disease, necessitating qualitative assessment or adjunctive immunostaining in addition to quantification of pathological inclusions. 2.1.2 Neuronal pathology FTLD-TDP type A demonstrates extensive cortical atrophy and proteinopathy with prominent involvement of the superficial cortical laminae. There is also evidence of deeper involvement, with many cases exhibiting short dystrophic neurites and compact NCIs in the deeper cortical laminae99. Cases with low levels of overall TDP pathology demonstrate inclusions in projection neurons and in oligodendrocytes of the orbital gyrus and gyrus rectus102. When the overall burden of TDP pathology increases during the disease progression, inclusions are found in the middle frontal gyrus, anterior cingulate gyrus, and insular cortex, as well as the superior and middle temporal gyri102. These findings provide support for an early involvement of the frontal and temporal lobes. This study however only included bvFTD cases and did not distinguish between type A and type B. To identify the type of neuron affected by neurodegeneration in FTLD-TDP, a recent study compared the postmortem tissue RNA-seq transcriptomes from the frontal cortex, temporal cortex, and cerebellum of 28 control and 30 FTLD-TDP cases. The analysis showed that neuronal loss in the cortex mainly concerns excitatory neurons103. Unfortunately, this study did not include pathological subclassification of FTLD-TDP cases in its analysis. As in PiD, VENs appear to exhibit selective vulnerability in FTLD-TDP104. Additionally, a population of pyramidal cells neighboring VENs, which likewise express the GABA receptor subunit theta (GABRQ) seem vulnerable in FTLD-TDP. The loss of GABRQ-expressing neurons appears to be correlated with the development of behavioural symptoms at least in FTLD-TDP and FTLD-FUS104. Among cases with a C9orf72 repeat expansion, type A cases appear to demonstrate particularly severe loss of VENs and neighboring GABRQ-expressing pyramidal cells104. FTLD-TDP cases with GRN mutations have also been shown to exhibit decreased densities of GABRQ-expressing cells104. Whether VENs are involved in sporadic cases of FTLD-TDP type A remains poorly understood. While some studies have examined sporadic bvFTD105,106, few of the cases examined were clearly type A. FTLD-TDP type A often affects the hippocampus, where the burden of disease can be quite severe. The pathological findings frequently meet criteria for hippocampal sclerosis, defined as “severe hippocampal neuronal loss and gliosis”99,107. In the hippocampal dentate gyrus, FTLD-TDP type A cases exhibit a moderate burden of compact NCIs, though they demonstrate a significantly lower burden of compact NCIs than type B or C cases99. Unlike other types of FTLD-TDP, type A cases also often exhibit NIIs in the dentate gyrus. Additionally, these cases consistently have delicate wispy threads in CA1, a change that is frequently associated with hippocampal sclerosis. This feature was found to be 100 % sensitive and specific for type A cases in one study99. Additional studies are needed to address whether these pathological features show selective predilection for specific hippocampal neuronal subpopulations. The basal ganglia are often involved in cases of FTLD-TDP type A. Long et al. have recently suggested that the degree of atrophy in the bilateral caudate and right putamen can distinguish between types A and B with high sensitivity and specificity108. These findings are consistent with previous research on the extracortical distribution of dystrophic neurites and neuronal inclusions in FTLD-TDP subtypes. Here, a significant difference was found between dystrophic neurite density in the putamen of type A cases when compared with type B109. Furthermore, both FTLD-GRN and FTLD-TDP type A without mutations in progranulin have been diagnosed on autopsy in cases of clinical CBS110, suggesting that involvement of the striatum can be widespread and clinically significant. Analysis of striatal neuron populations using anti-calcineurin antibodies has revealed marked loss of substance-P positive efferents to the substantia nigra and the globus pallidus pars interna111. Enkephalin-positive efferents to the globus pallidus pars externa were also affected, though not as severely, and ChAT-positive striatal interneurons were mostly unaffected111. The affected areas also exhibited proliferation of GFAP-positive astrocytes, and the severity of neuron loss was correlated with both the accumulation of phosphorylated TDP-43 inclusions and clinical cognitive symptoms111. While these findings have yet to be replicated in a large study powered to distinguish between subtypes of FTLD-TDP, they suggest a selective vulnerability of substance-P-positive striatal efferents. Thalamic atrophy is a relatively common finding in FTLD. MRI findings have suggested that FTLD-TDP cases exhibit greater thalamic involvement than FTLD-tau or FTLD-FUS112. In particular, FTLD-TDP type A cases demonstrate significant atrophy in most nuclei of the thalamus when compared to controls, with the possible exception of the ventral posterolateral, ventral medial, pulvinar, and medial geniculate nuclei, where only a trend has been demonstrated37. However, more research is necessary to replicate these findings at the histological level and to explore selectively vulnerable populations since only a minority of studies on FTLD-TDP include thalamic histopathological data113. Research into brainstem involvement in FTLD-TDP has been limited, and most studies do not explicitly distinguish between subtypes of FTLD-TDP. One study found evidence for dystrophic neurites and multiple types of NCI in the substantia nigra, without involvement of the hypoglossal nucleus99. Another small study found that the superior colliculus was involved in all subtypes of FTLD-TDP, and that the substantia nigra, red nucleus, and raphe nuclei also consistently exhibited TDP-43-positive inclusions114. More studies are needed to dissect the corresponding cellular details. The extent of cerebellar involvement in FTLD-TDP type A is currently uncertain. Although imaging studies of patients with FTLD-GRN fail to demonstrate significant cerebellar atrophy115, it is unclear whether this is also the case for sporadic FTLD-TDP type A cases. This may be partly explained by the lack of cerebellar TDP-43 accumulation apparent on histology even in very late stages of disease progression116. Together, the current information on vulnerable neuronal populations in FTLD-TDP type A points towards selective involvement of excitatory and GABRQ-expressing cortical neurons, substance P-secreting neurons, and to a lesser extent also enkephalin-positive striatal neurons. In the rest of the brain, the distribution of neuronal TDP-43 pathology has been well described, but the degree of subtype-specific neuronal loss relative to this pathology remains unexplored. 2.1.3 Glial involvement Astrocytic pathology has not been well-studied in FTLD-TDP type A. Some FTLD-TDP type A cases exhibit glial cytoplasmic inclusions, though they do not appear to be as abundant as in FTLD-TDP type B99,117. These glial cytoplasmic inclusions appear to localize primarily to oligodendrocytes in the white matter based on morphology and IHC evidence117. Other than the above, immunohistochemical evaluation of astroglial and oligodendroglial involvement in the brain has been largely neglected with the exception of the identification of increased GFAP reactivity in the basal ganglia in general FTLD-TDP111 and thalamus in FTLD-GRN118. Of promise, conventional IHC and mass spectrometry analysis of the insoluble proteome in FTLD-TDP cases has recently identified a unique pattern of astrocytic F-box protein 2 (FBXO2) expression specifically in type A cases. Further studies will however be required to clarify the proteomic alterations and distribution of these FBXO2-positive astrocytes119. Overall, the extent of macroglial involvement in FTLD-TDP type A and its significance for pathogenesis remains largely unexplored. Microglial activation, assessed using CD68 immunostaining, has been shown to be increased in the superficial cortical laminae I-III in FTLD-TDP type A43. While the genetic and histological FTLD-TDP classifications do not perfectly match, cases of FTLD with a GRN mutation have been shown to exhibit more superficial cortical microglial activation than cases with a C9orf72 expansion. This suggests that inherited forms of FLTD-TDP type A due to GRN mutations may involve greater microglial activation in affected grey matter regions than inherited type B or AB forms due to C9orf72 expansion120. The RNA-seq analysis described above also showed that increases in microglial and endothelial cell expression were highly correlated with neuronal loss103. The frontal and temporal white matter both display consistent microglial activation when assessed morphologically or by positivity for CD6842,43. Historically, it was hypothesized that cases with a GRN mutation may exhibit more microglial activation based on the inflammatory functions of progranulin. Yet, post-mortem studies have found no difference between FTLD-TDP cases with or without GRN mutations43. One study which included mostly type A cases but did not separately analyze results by subtype, found that microglial activation in the hippocampal white matter is significantly greater in FTLD-TDP cases than in controls121. In the CA1 region, however, no significant difference was found between FTLD-TDP cases and controls. In the dentate gyrus, the same study found that FTLD-TDP cases actually exhibited less microglial activation than controls121. In the thalamus of FTLD-GRN cases, Iba-1-positive microglia and GFAP-positive astrocytes were increased in number while myelination measured by myelin basic protein IHC was decreased118. To conclude, in FTLD-TDP type A microglial activation appears to largely mirror neuronal pathology and loss. There is however not enough available data to draw significant conclusions on the distribution and severity of macroglial involvement in FTLD-TDP type A and its potential pathophysiological relevance. 2.2 FTLD-TDP type B 2.2.1 General features FTLD-TDP type B is characterized by at least moderate numbers of NCIs throughout all layers of the cortex94. These inclusions usually have a diffuse granular morphology, in contrast to the compact elliptical or crescentic inclusions found in type A100 (Figure 3). Clinically, FTLD-TDP type B usually presents as bvFTD or as motor neuron disease plus FTD, with a significant clinical and genetic overlap between FTD and ALS94. There are both sporadic and genetic forms of FTLD-TDP type B, and the genetic causes usually overlap with genes implicated in the pathogenesis of ALS. FTLD-TDP types B or AB are most frequently associated with pathological C9orf72 hexanucleotide repeat expansions, and particularly prone to manifest with psychotic symptoms and motor neuron disease (MND)100. However, it is important to recognize that the association between type B pathology and psychotic symptoms is not entirely explained by the effect of C9orf72 expansions, as sporadic cases with type B pathology also display an increased frequency of psychotic symptoms96. Patients with C9orf72 mutations produce aggregation-prone dipeptide repeat (DPR) proteins including glycine-alanine (poly-GA), glycine-arginine (poly-GR), proline-alanine (poly-PA), proline-arginine (poly-PR), and glycine-proline (poly-GP), due to unconventional translation of the abnormal hexanucleotide repeat expansion in C9orf72122. In these patients, the DPRs are found throughout the brain, with the cerebellum exhibiting the greatest concentration of total and soluble DPRs123. This review will not explore region-specific expression of dipeptide repeat proteins as a definite positive association between DPR burden and disease-associated clinical symptoms has not been established. 2.2.2 Neuronal pathology The predominance of early behavioral symptoms in FTLD-TDP type B suggests that the orbitofrontal cortex and/or limbic structures are affected early during disease. Evidence to support this hypothesis comes from data-driven machine staging models116 and retrospective chart reviews after definitive diagnosis124. However, familial forms of FTLD-TDP can have different patterns of clinical and pathological progression, and these differences can be even more pronounced in comparison to sporadic cases23. As described for type A, a transcriptomic study102 showed evidence of selective loss of excitatory cortical neurons in FTLD-TDP, but this study did not include FTLD-TDP subtyping103. An earlier study from 1993 on cases with FTLD-TDP associated with ALS showed significant decreases in calbindin positivity without changes in expression of parvalbumin, suggesting that neurons expressing calbindin may be affected to a greater degree than neighboring neurons125. Yet, this study was performed before the discovery of TDP-43 and thus predated the modern classification of FTLD-TDP. Like FTLD-TDP type A cases, type B cases often involve VENs of the anterior cingulate cortex. As described by Nana et al., even early-stage cases appear to exhibit disproportionate cytoplasmic TDP-43 immunopositivity in VENs, and the accumulation of TDP-43 pathology in this neuronal population correlates with clinical severity126. Additionally, fork cells, which are found in the frontal insula alongside VENs, have been found to display a similar pattern of early neurodegeneration126. In both VENs and fork cells, cells displaying inclusions or nuclear TDP-43 depletion develop somatodendritic atrophy, suggesting direct neurotoxicity126. While some of the FTLD-TDP type B cases studied had C9orf72 repeat expansions, most were sporadic. Further evidence for the involvement of VENs in sporadic FTLD-TDP type B comes from earlier studies on sporadic bvFTD, in which many cases were either unclassified or type B105,106. In the hippocampus, FTLD-TDP type B cases demonstrate a greater burden of both compact and diffuse NCIs in the dentate gyrus and in the CA1 region of the hippocampus compared to FTLD-TDP type A99. However, FTLD-TDP type B cases do not demonstrate hippocampal NIIs, dystrophic neurites, or threads, and as a result, the total TDP-43 burden seen by IHC is lower than in FTLD-TDP type A cases in the CA1 region99. FTLD-TDP type B cases also demonstrate diffuse NCIs and glial cell inclusions in the basal ganglia and substantia nigra99,109. As discussed above, FTLD-TDP type B cases also appear to involve loss of substance-P positive striatal efferents, with milder involvement of enkephalin-positive efferents. Furthermore, striatal involvement may be less severe in FTLD-TDP type B cases than in type A cases108,111. Of note, all FTLD-TDP type B cases demonstrating this apparent selective neuronal vulnerability presented clinically as FTD-MND111. The thalamus appears to undergo minimal changes in most cases of FTLD-TDP type B. Only small numbers of thalamic NCIs tend to develop in type B cases109. Imaging studies demonstrate relatively minor focal atrophy in the lateral geniculate nucleus, ventral lateral, and mediodorsal nuclei of the thalamus37. C9orf72 repeat expansions however may predispose to more widespread thalamic atrophy, with greater involvement of the pulvinar37,127, but the exact cellular underpinnings of this change remain elusive. Brainstem pathology is a core feature of ALS, and limited evidence suggests that involvement of the brainstem may be more widespread in FTLD-TDP type B than in type A or C114,128,129. In addition, involvement of cranial nerve motor nuclei is a well documented feature of FTLD-TDP type B8. FTLD-TDP type B often features glial cytoplasmic inclusions in the medulla, in addition to widespread neuronal alterations in the hypoglossal nucleus23,99. The latter are significantly more pronounced in FTLD-TDP type B than in other FTLD-TDP types. Cerebellar TDP-43 pathology in FTLD-TDP is poorly documented, and the existing literature often does not stratify by histopathological subtype. Nonetheless, some clinically diagnosed cases of bvFTD or FTD-MND, with or without a C9orf72 repeat expansions, demonstrate mild but statistically significant atrophy in various regions of the cerebellum115,130. It is tempting to speculate that some of these cases may represent FTLD-TDP type B, but it is impossible to draw firm conclusions without autopsy confirmation. Histologically, no significant neuronal TDP-43 pathology or neurodegeneration has been seen in FTLD-TDP type B cases99,131, hence the molecular correlate substrate of cerebellar atrophy in FTLD-TDP type B remains unknown. Overall, current evidence suggests a selective vulnerability of cortical VENs, fork cells and motor neurons in FTLD-TDP type B. Preliminary data suggest that neocortical excitatory and calbindin-expressing neurons as well as substance-P positive striatal efferents are also affected. The selective neuronal vulnerability of other brain regions to TDP-43 pathology remains however largely unexplored. The relationship between TDP-43 pathology and loss of neuronal subtypes has only been established for VENs, fork cells and motor neurons. 2.2.3 Glial involvement Astrocytic pathology has not been well characterized in FTLD-TDP type B. White matter pathology is less pronounced in FTLD-TDP type B than in type A, and most TDP-43 localizes to oligodendrocytes117. In FTLD-TDP type B cases, activated microglia are distributed throughout all layers of the neocortex43. No significant differences have been found between sporadic FTLD-TDP and cases associated with a C9orf72 mutation43. In contrast to FTLD-TDP types A and C, FTLD-TDP type B cases and controls show similar microglial burden as assessed by CD68, Iba1, or CR3/43, according to pairwise comparisons between cases and controls42,43. However, a trend towards increased CD68 reactivity was identified in the frontal white matter compared to controls43, suggesting that differences in microglial activation may have been overlooked in published yet underpowered studies. In conclusion, additional human studies are warranted to evaluate the involvement of macroglia and microglia in FTLD-TDP type B and their potential pathophysiological roles. 2.3 FTLD-TDP type C 2.3.1 General features FTLD-TDP type C accounts for 25 % of FTLD-TDP cases8. It is characterized by the presence of long dystrophic neurites, which tend to be concentrated in the superficial cortical laminae but are also present in deeper cortical layers94,99 (Figure 3). In FTLD-TDP type C there are few neuronal cytoplasmic inclusions and very few neuronal intranuclear inclusions94,99. Clinically, this form of FTLD usually manifests as svPPA, and sometimes also as nfvPPA or bvFTD94. 2.3.2 Neuronal pathology Type C cases typically demonstrate asymmetric cortical atrophy predominating in the temporal lobe38. In affected regions, superficial dystrophic neurites are visible on IHC, which are usually long and thick, in contrast to the shorter, comma-shaped dystrophic neurites of FTLD-TDP type A cases94. It is unclear which type of neuron is most vulnerable to the development of these inclusions. For instance, VENs have not been studied in a cohort enriched for FTLD-TDP type C cases. In type FTLD-TDP type C cases, the dentate gyrus of the hippocampus exhibits compact NCIs that have been described as “Pick body-like” due to their uniformity99. The CA1 region does not generally contain NCIs, but does exhibit dystrophic neurites, which are usually not seen in FTLD-TDP type A or type B cases99. Notably, only a minority of cases demonstrate frank hippocampal sclerosis109. Like the hippocampus, the striatum displays dystrophic neurites99 and compact NCIs with round contours in FTLD-TDP type C99. The nucleus accumbens appears to accumulate a greater pathological burden than the dorsal striatum, though the mechanism for this apparent regional selectivity is unknown132. Riku et al. found that two out of five FTLD-TDP type C cases displayed a selective degeneration of striatal efferents as described above for FTLD-TDP type A. Caution is however warranted given the low number of FTLD type C cases studied111. In contrast to FTLD-TDP type A and type B cases, type C cases typically do not exhibit thalamic NCIs109. While one imaging study has found mild atrophy in the mediodorsal nucleus relative to controls, there was less thalamic atrophy than in any other pathological subtype of FTLD37. FTLD-TDP type C cases also exhibit only mild dystrophic neurites in the substantia nigra. As in type A, the hypoglossal nucleus is largely spared99. One study involving nine FTLD-TDP type C cases found no evidence of brainstem pathology in the midbrain, hypoglossal nucleus, or inferior olivary nucleus109, while another study involving five type C cases found TDP-43 deposits consistently in the superior colliculus, with occasional involvement of the inferior olivary nucleus and the red nucleus114. The cerebellum has not been systematically studied in FTLD-TDP type C. As discussed above for FTLD-TDP type A, some indirect evidence exists for cerebellar involvement FTLD-TDP in genera, but there have been no large studies examining differences between pathological subtypes, and the clinical significance of these findings remains unclear. Unlike FTLD-TDP type A and B cases, FTLD-TDP type C cases appear to show concentrated TDP-43 pathology in the neocortex, hippocampus, and anterior striatum. It is surprising that the identity of vulnerable neurons that develop the characteristic long ‘corkscrew’ neurite is still unknown more than a decade after the formal recognition of this pathological FTLD subtype. 2.3.3 Glial involvement It appears that glial inclusions, particularly subcortical white matter ones, are uncommon in FTLD-TDP type C117. It remains however unclear why TDP-43 does not tend to accumulate in cells of oligodendroglial morphology in FTLD-TDP type C cases, in contrast to the situation in types A and B. Microglial activation has been studied to a significant degree in FTLD-TDP type C. Here, it appears to follow a similar pattern to that observed in type A cases, with more activation in the superficial cortical laminae and predominant white matter microgliosis in the frontal lobe42,43. Caution is however warranted, since the corresponding studies included only five42 and seven43 cases with FTLD-TDP type C pathology. These limited data suggest that macroglia are involved in FTLD-TDP type C in a less prominent manner than in other FTLD-TDP subtypes. Further studies evaluating astrocytic and microglial reactivity as well as oligodendroglial loss throughout the brain of FTLD-TDP type C cases could provide powerful insights into this question. 3. Frontotemporal lobar degeneration with fused in sarcoma (FUS) pathology 3.1 General features About five percent of FTLD cases exhibit neither tau nor TDP-43 pathology, and many of these cases are positive for FUS on IHC8. Three phenotypes of FUS-positive, TDP-43-negative, tau-negative FTLD have been described: atypical frontotemporal lobar degeneration with ubiquitin inclusions (aFTLD-U), basophilic inclusion body disease (BIBD), and neuronal intermediate filament inclusion disease (NIFID)133. The most common subtype of FTLD-FUS, aFTLD-U, is characterized histologically by round, oval, or bean-shaped FUS-immunoreactive NCIs with a wide pattern of distribution134. These inclusions are also positive for Transportin 1, which mediates the nuclear import of FUS135. Additionally, aFTLD-U cases demonstrate long, thin, curved NIIs that have been described as “vermiform”136. BIBD cases exhibit severe striatal and nigral atrophy, with varying degrees of cortical atrophy corresponding to behavioural symptoms137,138. The disease gets its name from basophilic cytoplasmic inclusions that are negative for neurofilament and tau but positive for FUS and occasionally positive for ubiquitin or p62138. Colocalization immunofluorescence has shown that FUS accumulates in inclusions identified by hematoxylin and eosin staining and inclusions identified by p62 IHC. In addition, FUS also accumulates in many more inclusions that could not be detected using other methods than IHC138. For this reason, BIBD is now generally classified as a FUS proteinopathy, though there are still many open questions about its pathogenesis and presentation. NIFID cases also demonstrate involvement of various cortical and subcortical regions, and the observed NCIs and NIIs are heterogenous in their morphology and immunoreactivity139. The NCIs can be round, crescentic, annular, or tangle-like. In addition there are so-called “hyaline conglomerate inclusions” with a filamentous appearance and an eosinophilic core140. Both vermiform and round NIIs have been reported, and these appear to be more common in neurons that also exhibit round cytoplasmic inclusions139,140. These inclusions are immunoreactive for intermediate filament proteins and FUS, and colocalization of the latter has been confirmed using double-label immunofluorescence139. Nonetheless, some FTLD-FUS cases are immunoreactive for the intermediate filament alpha-internexin but not for FUS. Interestingly, one such case has been reported to exhibit both TDP-43 and alpha-internexin positivity141. Clinically, aFTLD with ubiquitin inclusions (aFTLD-U) typically manifests early in life as an unusual form of bvFTD that can involve obsessions, pica, ritualistic behavior, and hypersexuality133. Due to their rarity, other forms of FTLD-FUS have not been characterized as systematically, but case reports suggest that they are also likely to present with early-onset bvFTD or FTD-MND133,137,138,141,142. While stereotyped and repetitive behaviours are reported in some cases, these symptoms are not as consistent as in aFTLD-U. Conversely, prominent MND and parkinsonism seem to be more common in both BIBD and NIFID133,137,141, and BIBD more commonly involves memory impairment and apraxia133,137. 3.2 Neuronal pathology Cases of aFTLD-U demonstrate widespread NCIs throughout the frontal and temporal cortices143 and occasional NIIs in pyramidal neocortical neurons14. BIBD and NIFID cases also sometimes demonstrate neuronal inclusions in the frontal and temporal cortices137, but there are case reports of late-onset BIBD that preferentially affects the motor system, with minimal limbic or prefrontal involvement143. FTLD-FUS cases may also demonstrate selective involvement of VENs. While the sample size was small (n=8), one study found consistent and severe degeneration of GABRQ-positive neurons104. Moderate to severe hippocampal involvement is a common feature in aFTLD-U, with FUS-positive NCIs and vermiform NIIs in the granule cells of the dentate gyrus and, to a lesser extent, in the subiculum and CA1 regions14,133. In contrast, BIBD demonstrates less consistent involvement of the dentate granule cells and more consistent involvement of the pyramidal neurons134,136,138. Hippocampal inclusions can also be found in NIFID140,144, but in at least a subset of cases, inclusions in the hippocampus are not nearly as numerous as in the frontal lobe144. Severe striatal atrophy has been reported in all three major subtypes of FTLD-FUS136–138,140. In aFTLD-U, there are often crescentic NCIs and small numbers of NIIs136, and the striatum can exhibit varying degrees of involvement14,143,145. In BIBD, severe striatal atrophy is a consistent finding, and in some cases, striatal pathology is considerably more severe than cortical or hippocampal pathology138. Both caudate and putamen demonstrate severe neuronal loss, gliosis, and basophilic inclusions138, and early involvement of the striatum and the pyramidal motor system are more common in BIBD than in aFTLD-U143. As in BIBD, NIFID cases are thought to show severe, consistent striatal atrophy136,137, though a recent case series has presented numerous cases without evidence of atrophy141. The globus pallidus136–138,144 is also involved in FTLD-FUS, particularly in BIBD and NIFID but also in aFTLD-U14,136. However, some cases appear to exhibit only mild pallidal atrophy despite significant involvement of the striatum133,137, and there are some cases of aFTLD-U that involve extreme caudate atrophy with relative preservation of both putamen and globus pallidus133. In aFTLD-U cases, the thalamus is not a consistent site of pathology, but many cases show small numbers of NCIs and NIIs136,140. In contrast, BIBD appears to consistently demonstrate moderate to severe pathological burden in the thalamus, including non-compact collections of coarse granules and even occasional NIIs140. Finally, NIFID also shows relatively consistent involvement of the thalamus, though the level of pathology can vary considerably136,140,144. The brainstem also seems to be involved in aFTLD-U, with greater involvement of rostral regions143. However, many cases demonstrate only mild FUS-immunoreactive pathology in the pons and midbrain, including the substantia nigra but sparing the red nucleus140. In BIBD, there are basophilic, FUS-immunoreactive inclusions in the brainstem136–138, particularly in the pontine nuclei and the inferior olivary nucleus136. Indeed, along with the basal ganglia, the brainstem appears to be among the regions with the highest density of FUS-positive inclusions in BIBD137. Caudal regions of the brainstem display more pronounced pathology than rostral regions in BIBD143. Furthermore, lower motor neurons in the spinal cord are affected136,137. NIFID also shows more consistent and widespread brainstem pathology than aFTLD-U, including involvement of the locus coeruleus, the red nucleus, and the basis pontis140, though gross atrophy is not always apparent144. Much of the brainstem pathology presents as coarse granules rather than as compact inclusions139. Cerebellar involvement appears to be an uncommon finding in aFTLD-U, though some cases exhibit a small number of NCIs in the dentate nucleus136. Cerebellar involvement seems to be somewhat more common in BIBD, with NCIs and GCIs often present in the dentate nucleus137,138,143. Nonetheless, the cerebellar cortex is not generally affected143. NIFID cases also sometimes demonstrate neuronal loss and FUS-immunoreactive pathology in the cerebellum136,139. However, this does not appear to be a consistent finding144, and there are fewer FUS-immunoreactive inclusions in the cerebellum than in virtually any other grey matter region that has been subjected to study134. In summary, the distribution of FUS-immunoreactive pathology has been described in all three types of FTLD-FUS. However, only VENs and GABRQ+ neuronal subtypes have been proven to be affected by the pathological inclusions. Further subtype-specific studies of neuronal loss and correlative studies with FUS inclusions in each FTLD-FUS subtype are needed to understand the pathogenic mechanisms of disease in these entities. 3.3 Glial involvement All three major forms of FTLD-FUS are associated with glial cytoplasmic inclusions in the white matter14,134,139,140. These inclusions are typically oval or flame-shaped134. Similar inclusions have been identified in ALS with a FUS mutation145–147. Double labelling has demonstrated that the inclusions localize to oligodendrocytes145. Based on these findings and the morphological characteristics of the involved cells in FTLD-FUS, it seems reasonable to conclude that the GCIs represent pathological involvement of oligodendrocytes. The distribution and protein expression of affected cells have however not been systematically studied. In one study comparing microglial activation across subtypes of FTLD, FTLD-FUS cases did not appear to demonstrate greater grey matter microglial activation than controls. Yet, only four cases were included, and all cases were aFTLD-U42. In the white matter, there appears to be a possible trend toward increased microglial activation compared to controls in frontal and temporal white matter, but a statistically significant difference has not been demonstrated42. Conclusion There is clear evidence for selective region- and cell-type specific vulnerability in all forms of FTLD, which helps explain the distinct clinical and pathological features associated with each subtype. Where there is evidence of selective neuronal vulnerability, such as VENs in PiD and FTLD-TDP or striatal substance-P-positive efferents in FTLD-TDP, further characterization of the changes to those populations and comparisons to unaffected neuronal types would be of value. Where there is little evidence on selectively vulnerable populations, such as in FTLD-FUS, systematic attempts to identify and characterize vulnerable cell types could help focus further research. As the scientific community has learned from attempts to understand other neurodegenerative conditions, abnormal protein inclusions are excellent markers for disease classification and diagnosis but are unlikely to be the sole pathophysiological factor at play. This highlights the importance of systematic studies to understand selective vulnerability of both neurons and glia and the precise distribution of these cell types in FTLD. When a selectively vulnerable population is identified, exploration of associated glia, synaptic inputs, and projections can provide mechanistic information beyond that afforded by characterization of isolated affected neurons. This review suggests a number of areas for further research, including the evaluation of neuronal subtype-specific vulnerability; the significance and distribution of oligodendroglial and astrocytic pathology; the relationship between activated microglia and vulnerable neuronal populations; and the relative importance of cell-autonomous versus non-cell-autonomous mechanisms of neurodegeneration. This latter aspect has not been extensively evaluated in humans. Closing the knowledge gap on some of these areas may identify pathological processes amenable to therapeutic intervention, which will likely be disease specific as the existing literature suggests. Funding statement VHR is supported by the Vancouver Research Institute Mentored Clinician-Scientist Award. Conflict of interest The authors declare no conflict of interest. References 1. Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci MN. 2011;45(3):330-335. https://doi.org/10.1007/s12031-011-9538-y 2. Coyle-Gilchrist ITS, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology . 2016;86(18):1736-1743. https://doi.org/10.1212/WNL.0000000000002638 3. Woollacott IOC, Rohrer JD. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem . 2016;138 Suppl 1:6-31. https://doi.org/10.1111/jnc.13654 4. Kertesz A, McMonagle P, Jesso S. Extrapyramidal syndromes in frontotemporal degeneration. J Mol Neurosci MN . 2011;45(3):336-342. https://doi.org/10.1007/s12031-011-9616-1 5. Beber BC, Chaves MLF. Evaluation of patients with behavioral and cognitive complaints: misdiagnosis in frontotemporal dementia and Alzheimer’s disease. Dement Neuropsychol . 2013;7(1):60-65. https://doi.org/10.1590/S1980-57642013DN70100010 6. Ossenkoppele R, Pijnenburg YAL, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain J Neurol . 2015;138(Pt 9):2732-2749. https://doi.org/10.1093/brain/awv191 7. Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain J Neurol . 2017;140(12):3329-3345. https://doi.org/10.1093/brain/awx254 8. Love S, Perry A, Ironside J, Budka H. Greenfield’s Neuropathology - Two Volume Set. CRC Press; 2018. http://books.google.ca/books?id=y4V7DwAAQBAJ&printsec=frontcover&dq=greenfields+neuropathology+9th+ed&hl=&cd=1&source=gbs_api 9. Clark AW, White CL, Manz HJ, et al. Primary degenerative dementia without Alzheimer pathology. Can J Neurol Sci J Can Sci Neurol . 1986;13(4 Suppl):462-470. https://doi.org/10.1017/s0317167100037136 10. Love S, Saitoh T, Quijada S, Cole GM, Terry RD. Alz-50, ubiquitin and tau immunoreactivity of neurofibrillary tangles, Pick bodies and Lewy bodies. J Neuropathol Exp Neurol . 1988;47(4):393-405. https://doi.org/10.1097/00005072-198807000-00001 11. Pollock NJ, Mirra SS, Binder LI, Hansen LA, Wood JG. Filamentous aggregates in Pick’s disease, progressive supranuclear palsy, and Alzheimer’s disease share antigenic determinants with microtubule-associated protein, tau. Lancet Lond Engl . 1986;2(8517):1211. https://doi.org/10.1016/s0140-6736(86)92212-9 12. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science . 2006;314(5796):130-133. https://doi.org/10.1126/science.1134108 13. Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun . 2006;351(3):602-611. https://doi.org/10.1016/j.bbrc.2006.10.093 14. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain J Neurol . 2009;132(Pt 11):2922-2931. https://doi.org/10.1093/brain/awp214 15. The FReJA Consortium, Urwin H, Josephs KA, et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol (Berl) . 2010;120(1):33-41. https://doi.org/10.1007/s00401-010-0698-6 16. Ferrer I, López-González I, Carmona M, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol . 2014;73(1):81-97. https://doi.org/10.1097/NEN.0000000000000030 17. Mackenzie IRA, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol (Berl) . 2006;112(5):539-549. https://doi.org/10.1007/s00401-006-0138-9 18. Sampathu DM, Neumann M, Kwong LK, et al. Pathological Heterogeneity of Frontotemporal Lobar Degeneration with Ubiquitin-Positive Inclusions Delineated by Ubiquitin Immunohistochemistry and Novel Monoclonal Antibodies. Am J Pathol . 2006;169(4):1343-1352. https://doi.org/10.2353/ajpath.2006.060438 19. Leng K, Li E, Eser R, et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci . 2021;24(2):276-287. https://doi.org/10.1038/s41593-020-00764-7 20. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci . 2017;18(2):101-113. https://doi.org/10.1038/nrn.2016.178 21. Philips T, Rothstein JD. Glial cells in amyotrophic lateral sclerosis. Exp Neurol . 2014;262 Pt B:111-120. https://doi.org/10.1016/j.expneurol.2014.05.015 22. Boillée S, Yamanaka K, Lobsiger CS, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science . 2006;312(5778):1389-1392. https://doi.org/10.1126/science.1123511 23. Mackenzie IRA, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem . 2016;138 Suppl 1:54-70. https://doi.org/10.1111/jnc.13588 24. Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci MN . 2011;45(3):384-389. https://doi.org/10.1007/s12031-011-9589-0 25. Irwin DJ, Brettschneider J, McMillan CT, et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol . 2016;79(2):272-287. https://doi.org/10.1002/ana.24559 26. Dickson DW. Pick’s disease: a modern approach. Brain Pathol Zurich Switz . 1998;8(2):339-354. https://doi.org/10.1111/j.1750-3639.1998.tb00158.x 27. Hogg M, Grujic ZM, Baker M, et al. The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta Neuropathol (Berl) . 2003;106(4):323-336. https://doi.org/10.1007/s00401-003-0734-x 28. Arai T, Ikeda K, Akiyama H, et al. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol (Berl) . 2001;101(2):167-173. https://doi.org/10.1007/s004010000283 29. Schofield E, Kersaitis C, Shepherd CE, Kril JJ, Halliday GM. Severity of gliosis in Pick’s disease and frontotemporal lobar degeneration: tau-positive glia differentiate these disorders. Brain J Neurol . 2003;126(Pt 4):827-840. https://doi.org/10.1093/brain/awg085 30. Ohm DT, Cousins KAQ, Xie SX, et al. Signature laminar distributions of pathology in frontotemporal lobar degeneration. Acta Neuropathol (Berl) . 2022;143(3):363-382. https://doi.org/10.1007/s00401-021-02402-3 31. Seeley WW. Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr Opin Neurol . 2008;21(6):701-707. https://doi.org/10.1097/WCO.0b013e3283168e2d 32. Seeley WW, Carlin DA, Allman JM, et al. Early frontotemporal dementia targets neurons unique to apes and humans. Ann Neurol . 2006;60(6):660-667. https://doi.org/10.1002/ana.21055 33. Broe M, Kril J, Halliday GM. Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain J Neurol . 2004;127(Pt 10):2214-2220. https://doi.org/10.1093/brain/awh250 34. Kovacs GG, Rozemuller AJM, van Swieten JC, et al. Neuropathology of the hippocampus in FTLD-Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol Appl Neurobiol . 2013;39(2):166-178. https://doi.org/10.1111/j.1365-2990.2012.01272.x 35. Kawles A, Minogue G, Zouridakis A, et al. Differential vulnerability of the dentate gyrus to tauopathies in dementias. Acta Neuropathol Commun . 2023;11(1):1. https://doi.org/10.1186/s40478-022-01485-7 36. Stefanits H, Wesseling C, Kovacs GG. Loss of Calbindin immunoreactivity in the dentate gyrus distinguishes Alzheimer’s disease from other neurodegenerative dementias. Neurosci Lett . 2014;566:137-141. https://doi.org/10.1016/j.neulet.2014.02.026 37. Bocchetta M, Iglesias JE, Neason M, Cash DM, Warren JD, Rohrer JD. Thalamic nuclei in frontotemporal dementia: Mediodorsal nucleus involvement is universal but pulvinar atrophy is unique to C9orf72. Hum Brain Mapp . 2020;41(4):1006-1016. https://doi.org/10.1002/hbm.24856 38. Rohrer JD, Lashley T, Schott JM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain J Neurol . 2011;134(Pt 9):2565-2581. https://doi.org/10.1093/brain/awr198 39. Braak E, Arai K, Braak H. Cerebellar involvement in Pick’s disease: affliction of mossy fibers, monodendritic brush cells, and dentate projection neurons. Exp Neurol . 1999;159(1):153-163. https://doi.org/10.1006/exnr.1999.7131 40. de Wit NM, den Hoedt S, Martinez-Martinez P, Rozemuller AJ, Mulder MT, de Vries HE. Astrocytic ceramide as possible indicator of neuroinflammation. J Neuroinflammation . 2019;16(1):48. https://doi.org/10.1186/s12974-019-1436-1 41. Feany MB, Mattiace LA, Dickson DW. Neuropathologic overlap of progressive supranuclear palsy, Pick’s disease and corticobasal degeneration. J Neuropathol Exp Neurol . 1996;55(1):53-67. https://doi.org/10.1097/00005072-199601000-00006 42. Woollacott IOC, Toomey CE, Strand C, et al. Microglial burden, activation and dystrophy patterns in frontotemporal lobar degeneration. J Neuroinflammation . 2020;17(1):234. https://doi.org/10.1186/s12974-020-01907-0 43. Lant SB, Robinson AC, Thompson JC, et al. Patterns of microglial cell activation in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol . 2014;40(6):686-696. https://doi.org/10.1111/nan.12092 44. Rebeiz JJ, Kolodny EH, Richardson EP. Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol . 1968;18(1):20-33. https://doi.org/10.1001/archneur.1968.00470310034003 45. Burrell JR, Hodges JR, Rowe JB. Cognition in corticobasal syndrome and progressive supranuclear palsy: a review. Mov Disord Off J Mov Disord Soc . 2014;29(5):684-693. https://doi.org/10.1002/mds.25872 46. Kouri N, Whitwell JL, Josephs KA, Rademakers R, Dickson DW. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol . 2011;7(5):263-272. https://doi.org/10.1038/nrneurol.2011.43 47. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology . 2013;80(5):496-503. https://doi.org/10.1212/WNL.0b013e31827f0fd1 48. Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol . 2011;70(2):327-340. https://doi.org/10.1002/ana.22424 49. Ling H, O’Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain J Neurol . 2010;133(Pt 7):2045-2057. https://doi.org/10.1093/brain/awq123 50. Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol . 1999;246 Suppl 2:II6-15. https://doi.org/10.1007/BF03161076 51. Dickson DW, Bergeron C, Chin SS, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol . 2002;61(11):935-946. https://doi.org/10.1093/jnen/61.11.935 52. Boxer AL, Geschwind MD, Belfor N, et al. Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol . 2006;63(1):81-86. https://doi.org/10.1001/archneur.63.1.81 53. Sakae N, Santos OA, Pedraza O, et al. Clinical and pathologic features of cognitive-predominant corticobasal degeneration. Neurology . 2020;95(1):e35-e45. https://doi.org/10.1212/WNL.0000000000009734 54. Kłodowska-Duda G, Słowiński J, Opala G, et al. Corticobasal degeneration -- clinico-pathological considerations. Folia Neuropathol. 2006;44(4):257-264. PMID: 17183452 55. Yoshida M. Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathol Off J Jpn Soc Neuropathol . 2014;34(6):555-570. https://doi.org/10.1111/neup.12143 56. Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol . 2001;60(6):647-657. https://doi.org/10.1093/jnen/60.6.647 57. Kaasinen V, Gardberg M, Röyttä M, Seppänen M, Päivärinta M. Normal dopamine transporter SPECT in neuropathologically confirmed corticobasal degeneration. J Neurol . 2013;260(5):1410-1411. https://doi.org/10.1007/s00415-013-6886-2 58. Hattori M, Hashizume Y, Yoshida M, et al. Distribution of astrocytic plaques in the corticobasal degeneration brain and comparison with tuft-shaped astrocytes in the progressive supranuclear palsy brain. Acta Neuropathol (Berl) . 2003;106(2):143-149. https://doi.org/10.1007/s00401-003-0711-4 59. Oyanagi K, Tsuchiya K, Yamazaki M, Ikeda K. Substantia nigra in progressive supranuclear palsy, corticobasal degeneration, and parkinsonism-dementia complex of Guam: specific pathological features. J Neuropathol Exp Neurol . 2001;60(4):393-402. https://doi.org/10.1093/jnen/60.4.393 60. Piao YS, Hayashi S, Wakabayashi K, et al. Cerebellar cortical tau pathology in progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol (Berl) . 2002;103(5):469-474. https://doi.org/10.1007/s00401-001-0488-2 61. Vogel H, Butcher EC, Picker LJ. H-CAM expression in the human nervous system: evidence for a role in diverse glial interactions. J Neurocytol . 1992;21(5):363-373. https://doi.org/10.1007/bf01191704 62. Ikeda K, Akiyama H, Arai T, Nishimura T. Glial tau pathology in neurodegenerative diseases: their nature and comparison with neuronal tangles. Neurobiol Aging . 1998;19(1 Suppl):S85-91. https://doi.org/10.1016/s0197-4580(98)00034-7 63. Briel N, Pratsch K, Roeber S, Arzberger T, Herms J. Contribution of the astrocytic tau pathology to synapse loss in progressive supranuclear palsy and corticobasal degeneration. Brain Pathol Zurich Switz . 2021;31(4):e12914. https://doi.org/10.1111/bpa.12914 64. Nishida N, Yoshida K, Hata Y, Arai Y, Kinoshita K. Pathological features of preclinical or early clinical stages of corticobasal degeneration: a comparison with advanced cases. Neuropathol Appl Neurobiol . 2015;41(7):893-905. https://doi.org/10.1111/nan.12229 65. Ling H, Kovacs GG, Vonsattel JPG, et al. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain J Neurol . 2016;139(Pt 12):3237-3252. https://doi.org/10.1093/brain/aww256 66. Kobylecki C, Mann DM. Presymptomatic anterior frontal involvement in corticobasal degeneration. Brain J Neurol . 2016;139(Pt 12):3059-3062. https://doi.org/10.1093/brain/aww267 67. Yoo D, Park SH, Yu S, Ahn TB. An autopsy-proven case of Corticobasal degeneration heralded by Pontine infarction. BMC Neurol . 2021;21(1):148. https://doi.org/10.1186/s12883-021-02178-9 68. Chung DEC, Roemer S, Petrucelli L, Dickson DW. Cellular and pathological heterogeneity of primary tauopathies. Mol Neurodegener . 2021;16(1):57. https://doi.org/10.1186/s13024-021-00476-x 69. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord Off J Mov Disord Soc . 2017;32(6):853-864. https://doi.org/10.1002/mds.26987 70. Josephs KA, Katsuse O, Beccano-Kelly DA, et al. Atypical progressive supranuclear palsy with corticospinal tract degeneration. J Neuropathol Exp Neurol . 2006;65(4):396-405. https://doi.org/10.1097/01.jnen.0000218446.38158.61 71. Hauw JJ, Verny M, Delaère P, Cervera P, He Y, Duyckaerts C. Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer’s disease and aging. Neurosci Lett . 1990;119(2):182-186. https://doi.org/10.1016/0304-3940(90)90829-x 72. Lagarde J, Valabrègue R, Corvol JC, et al. Are frontal cognitive and atrophy patterns different in PSP and bvFTD? A comparative neuropsychological and VBM study. PloS One . 2013;8(11):e80353. https://doi.org/10.1371/journal.pone.0080353 73. Cordato NJ, Pantelis C, Halliday GM, et al. Frontal atrophy correlates with behavioural changes in progressive supranuclear palsy. Brain J Neurol . 2002;125(Pt 4):789-800. https://doi.org/10.1093/brain/awf082 74. Sakae N, Josephs KA, Litvan I, et al. Neuropathologic basis of frontotemporal dementia in progressive supranuclear palsy. Mov Disord Off J Mov Disord Soc . 2019;34(11):1655-1662. https://doi.org/10.1002/mds.27816 75. Halliday GM, Macdonald V, Henderson JM. A comparison of degeneration in motor thalamus and cortex between progressive supranuclear palsy and Parkinson’s disease. Brain J Neurol . 2005;128(Pt 10):2272-2280. https://doi.org/10.1093/brain/awh596 76. Stejskalova Z, Rohan Z, Rusina R, et al. Pyramidal system involvement in progressive supranuclear palsy - a clinicopathological correlation. BMC Neurol . 2019;19(1):42. https://doi.org/10.1186/s12883-019-1270-1 77. Armstrong RA, Lantos PL, Cairns NJ. Hippocampal pathology in progressive supranuclear palsy (PSP): a quantitative study of 8 cases. Clin Neuropathol . 2009;28(1):46-53. https://doi.org/10.5414/npp28046 78. Kovacs GG, Lukic MJ, Irwin DJ, et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol (Berl) . 2020;140(2):99-119. https://doi.org/10.1007/s00401-020-02158-2 79. Levy R, Ruberg M, Herrero MT, et al. Alterations of GABAergic neurons in the basal ganglia of patients with progressive supranuclear palsy: an in situ hybridization study of GAD67 messenger RNA. Neurology . 1995;45(1):127-134. https://doi.org/10.1212/wnl.45.1.127 80. Kasashima S, Oda Y. Cholinergic neuronal loss in the basal forebrain and mesopontine tegmentum of progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol (Berl) . 2003;105(2):117-124. https://doi.org/10.1007/s00401-002-0621-x 81. Henderson JM, Carpenter K, Cartwright H, Halliday GM. Loss of thalamic intralaminar nuclei in progressive supranuclear palsy and Parkinson’s disease: clinical and therapeutic implications. Brain J Neurol . 2000;123 ( Pt 7):1410-1421. https://doi.org/10.1093/brain/123.7.1410 82. Hardman CD, Halliday GM, McRitchie DA, Cartwright HR, Morris JG. Progressive supranuclear palsy affects both the substantia nigra pars compacta and reticulata. Exp Neurol . 1997;144(1):183-192. https://doi.org/10.1006/exnr.1997.6415 83. Murphy KE, Karaconji T, Hardman CD, Halliday GM. Excessive dopamine neuron loss in progressive supranuclear palsy. Mov Disord Off J Mov Disord Soc . 2008;23(4):607-610. https://doi.org/10.1002/mds.21907 84. Zemel D, Gritton H, Cheung C, Shankar S, Kramer M, Han X. Dopamine depletion selectively disrupts interactions between striatal neuron subtypes and LFP oscillations. Cell Rep . 2022;38(3):110265. https://doi.org/10.1016/j.celrep.2021.110265 85. Sébille SB, Rolland AS, Faillot M, et al. Normal and pathological neuronal distribution of the human mesencephalic locomotor region. Mov Disord Off J Mov Disord Soc . 2019;34(2):218-227. https://doi.org/10.1002/mds.27578 86. Kaalund SS, Passamonti L, Allinson KSJ, et al. Locus coeruleus pathology in progressive supranuclear palsy, and its relation to disease severity. Acta Neuropathol Commun . 2020;8(1):11. https://doi.org/10.1186/s40478-020-0886-0 87. Ando S, Kanazawa M, Onodera O. Progressive Supranuclear Palsy with Predominant Cerebellar Ataxia. J Mov Disord . 2020;13(1):20-26. https://doi.org/10.14802/jmd.19061 88. Togo T, Dickson DW. Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol (Berl) . 2002;104(4):398-402. https://doi.org/10.1007/s00401-002-0569-x 89. Ahmed Z, Asi YT, Lees AJ, Revesz T, Holton JL. Identification and quantification of oligodendrocyte precursor cells in multiple system atrophy, progressive supranuclear palsy and Parkinson’s disease. Brain Pathol Zurich Switz . 2013;23(3):263-273. https://doi.org/10.1111/j.1750-3639.2012.00637.x 90. Zhukareva V, Joyce S, Schuck T, et al. Unexpected abundance of pathological tau in progressive supranuclear palsy white matter. Ann Neurol . 2006;60(3):335-345. https://doi.org/10.1002/ana.20916 91. Rohan Z, Milenkovic I, Lutz MI, Matej R, Kovacs GG. Shared and Distinct Patterns of Oligodendroglial Response in α-Synucleinopathies and Tauopathies. J Neuropathol Exp Neurol . 2016;75(12):1100-1109. https://doi.org/10.1093/jnen/nlw087 92. Santpere G, Ferrer I. Delineation of early changes in cases with progressive supranuclear palsy-like pathology. Astrocytes in striatum are primary targets of tau phosphorylation and GFAP oxidation. Brain Pathol Zurich Switz . 2009;19(2):177-187. https://doi.org/10.1111/j.1750-3639.2008.00173.x 93. Armstrong RA, Cairns NJ. Spatial patterns of the tau pathology in progressive supranuclear palsy. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol . 2013;34(3):337-344. https://doi.org/10.1007/s10072-012-1006-0 94. Mackenzie IRA, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol (Berl) . 2011;122(1):111-113. https://doi.org/10.1007/s00401-011-0845-8 95. Lee EB, Porta S, Baer GM, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol (Berl) . Published online January 2017:1-14. https://doi.org/10.1007/s00401-017-1679-9 96. Hirsch-Reinshagen V, Hercher C, Vila-Rodriguez F, et al. Psychotic symptoms in frontotemporal dementia with TDP-43 tend to be associated with type B pathology. Neuropathol Appl Neurobiol . 2023;49(4):e12921. https://doi.org/10.1111/nan.12921 97. Chen-Plotkin AS, Martinez-Lage M, Sleiman PMA, et al. Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol . 2011;68(4):488-497. https://doi.org/10.1001/archneurol.2011.53 98. Mao Q, Zheng X, Gefen T, et al. FTLD-TDP With and Without GRN Mutations Cause Different Patterns of CA1 Pathology. J Neuropathol Exp Neurol . 2019;78(9):844-853. https://doi.org/10.1093/jnen/nlz059 99. Mackenzie IR, Neumann M. Subcortical TDP-43 pathology patterns validate cortical FTLD-TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol (Berl) . 2020;139(1):83-98. https://doi.org/10.1007/s00401-019-02070-4 100. Neumann M, Lee EB, Mackenzie IR. Frontotemporal Lobar Degeneration TDP-43-Immunoreactive Pathological Subtypes: Clinical and Mechanistic Significance. Adv Exp Med Biol . 2021;1281:201-217. https://doi.org/10.1007/978-3-030-51140-1_13 101. Yousef A, Robinson JL, Irwin DJ, et al. Neuron loss and degeneration in the progression of TDP-43 in frontotemporal lobar degeneration. Acta Neuropathol Commun . 2017;5(1):68. https://doi.org/10.1186/s40478-017-0471-3 102. Brettschneider J, Del Tredici K, Irwin DJ, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol (Berl) . 2014;127(3):423-439. https://doi.org/10.1007/s00401-013-1238-y 103. Hasan R, Humphrey J, Bettencourt C, et al. Transcriptomic analysis of frontotemporal lobar degeneration with TDP-43 pathology reveals cellular alterations across multiple brain regions. Acta Neuropathol (Berl) . 2022;143(3):383-401. https://doi.org/10.1007/s00401-021-02399-9 104. Gami-Patel P, van Dijken I, van Swieten JC, et al. Von Economo neurons are part of a larger neuronal population that are selectively vulnerable in C9orf72 frontotemporal dementia. Neuropathol Appl Neurobiol . 2019;45(7):671-680. https://doi.org/10.1111/nan.12558 105. Santillo AF, Englund E. Greater loss of von Economo neurons than loss of layer II and III neurons in behavioral variant frontotemporal dementia. Am J Neurodegener Dis. 2014;3(2):64-71. PMID: 25232511 106. Kim EJ, Sidhu M, Gaus SE, et al. Selective frontoinsular von Economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb Cortex N Y N 1991 . 2012;22(2):251-259. https://doi.org/10.1093/cercor/bhr004 107. Sieben A, Van Langenhove T, Vermeiren Y, et al. Hippocampal Sclerosis in Frontotemporal Dementia: When Vascular Pathology Meets Neurodegeneration. J Neuropathol Exp Neurol . 2021;80(4):313-324. https://doi.org/10.1093/jnen/nlab010 108. Long Z, Irish M, Hodges JR, Halliday G, Piguet O, Burrell JR. Amyotrophic lateral sclerosis features predict TDP-43 pathology in frontotemporal lobar degeneration. Neurobiol Aging . 2021;107:11-20. https://doi.org/10.1016/j.neurobiolaging.2021.07.004 109. Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol (Berl) . 2009;118(3):349-358. https://doi.org/10.1007/s00401-009-0547-7 110. Tartaglia MC, Sidhu M, Laluz V, et al. Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol (Berl) . 2010;119(3):365-374. https://doi.org/10.1007/s00401-009-0605-1 111. Riku Y, Watanabe H, Yoshida M, et al. Marked Involvement of the Striatal Efferent System in TAR DNA-Binding Protein 43 kDa-Related Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. J Neuropathol Exp Neurol . 2016;75(8):801-811. https://doi.org/10.1093/jnen/nlw053 112. Bocchetta M, Gordon E, Cardoso MJ, et al. Thalamic atrophy in frontotemporal dementia - Not just a C9orf72 problem. NeuroImage Clin . 2018;18:675-681. https://doi.org/10.1016/j.nicl.2018.02.019 113. McKenna MC, Lope J, Bede P, Tan EL. Thalamic pathology in frontotemporal dementia: Predilection for specific nuclei, phenotype-specific signatures, clinical correlates, and practical relevance. Brain Behav . 2023;13(2):e2881. https://doi.org/10.1002/brb3.2881 114. Grinberg LT, Rueb U, Heinsen H. Brainstem: neglected locus in neurodegenerative diseases. Front Neurol . 2011;2:42. https://doi.org/10.3389/fneur.2011.00042 115. Whitwell JL, Weigand SD, Boeve BF, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain . 2012;135(3):794-806. https://doi.org/10.1093/brain/aws001 116. Young AL, Vogel JW, Robinson JL, et al. Data-driven neuropathological staging and subtyping of TDP-43 proteinopathies. Brain J Neurol . 2023;146(7):2975-2988. https://doi.org/10.1093/brain/awad145 117. Neumann M, Kwong LK, Truax AC, et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol . 2007;66(3):177-183. https://doi.org/10.1097/01.jnen.0000248554.45456.58 118. Marsan E, Velmeshev D, Ramsey A, et al. Astroglial toxicity promotes synaptic degeneration in the thalamocortical circuit in frontotemporal dementia with GRN mutations. J Clin Invest . 2023;133(6):e164919. https://doi.org/10.1172/JCI164919 119. Laferrière F, Maniecka Z, Pérez-Berlanga M, et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci . 2019;22(1):65-77. https://doi.org/10.1038/s41593-018-0294-y 120. Sakae N, Roemer SF, Bieniek KF, et al. Microglia in frontotemporal lobar degeneration with progranulin or C9ORF72 mutations. Ann Clin Transl Neurol . 2019;6(9):1782-1796. https://doi.org/10.1002/acn3.50875 121. Taipa R, Brochado P, Robinson A, et al. Patterns of Microglial Cell Activation in Alzheimer Disease and Frontotemporal Lobar Degeneration. Neurodegener Dis . 2017;17(4-5):145-154. https://doi.org/10.1159/000457127 122. Mackenzie IRA, Frick P, Grässer FA, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol (Berl) . Published online September 2015. https://doi.org/10.1007/s00401-015-1476-2 123. Quaegebeur A, Glaria I, Lashley T, Isaacs AM. Soluble and insoluble dipeptide repeat protein measurements in C9orf72-frontotemporal dementia brains show regional differential solubility and correlation of poly-GR with clinical severity. Acta Neuropathol Commun . 2020;8(1):184. https://doi.org/10.1186/s40478-020-01036-y 124. Kawakami I, Arai T, Shinagawa S, Niizato K, Oshima K, Ikeda M. Distinct early symptoms in neuropathologically proven frontotemporal lobar degeneration. Int J Geriatr Psychiatry . 2021;36(1):38-45. https://doi.org/10.1002/gps.5387 125. Ferrer I, Tuñón T, Serrano MT, et al. Calbindin D-28k and parvalbumin immunoreactivity in the frontal cortex in patients with frontal lobe dementia of non-Alzheimer type associated with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry . 1993;56(3):257-261. https://doi.org/10.1136/jnnp.56.3.257 126. Nana AL, Sidhu M, Gaus SE, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol (Berl) . 2019;137(1):27-46. https://doi.org/10.1007/s00401-018-1942-8 127. Schönecker S, Neuhofer C, Otto M, et al. Atrophy in the Thalamus But Not Cerebellum Is Specific for C9orf72 FTD and ALS Patients - An Atlas-Based Volumetric MRI Study. Front Aging Neurosci . 2018;10:45. https://doi.org/10.3389/fnagi.2018.00045 128. Bede P, Chipika RH, Finegan E, et al. Brainstem pathology in amyotrophic lateral sclerosis and primary lateral sclerosis: A longitudinal neuroimaging study. NeuroImage Clin . 2019;24:102054. https://doi.org/10.1016/j.nicl.2019.102054 129. Cooper-Knock J, Hewitt C, Highley JR, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain J Neurol . 2012;135(Pt 3):751-764. https://doi.org/10.1093/brain/awr365 130. Bocchetta M, Todd EG, Tse NY, et al. Thalamic and Cerebellar Regional Involvement across the ALS-FTD Spectrum and the Effect of C9orf72. Brain Sci . 2022;12(3):336. https://doi.org/10.3390/brainsci12030336 131. Hsiung GYR, DeJesus-Hernandez M, Feldman HH, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain J Neurol . 2012;135(Pt 3):709-722. https://doi.org/10.1093/brain/awr354 132. Halabi C, Halabi A, Dean DL, et al. Patterns of striatal degeneration in frontotemporal dementia. Alzheimer Dis Assoc Disord . 2013;27(1):74-83. https://doi.org/10.1097/WAD.0b013e31824a7df4 133. Snowden JS, Hu Q, Rollinson S, et al. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol (Berl) . 2011;122(1):99-110. https://doi.org/10.1007/s00401-011-0816-0 134. Mackenzie IRA, Neumann M. Fused in Sarcoma Neuropathology in Neurodegenerative Disease. Cold Spring Harb Perspect Med . 2017;7(12):a024299. https://doi.org/10.1101/cshperspect.a024299 135. Neumann M, Valori CF, Ansorge O, et al. Transportin 1 accumulates specifically with FET proteins but no other transportin cargos in FTLD-FUS and is absent in FUS inclusions in ALS with FUS mutations. Acta Neuropathol (Berl) . 2012;124(5):705-716. https://doi.org/10.1007/s00401-012-1020-6 136. Mackenzie IRA, Munoz DG, Kusaka H, et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol (Berl) . 2011;121(2):207-218. https://doi.org/10.1007/s00401-010-0764-0 137. Yokota O, Tsuchiya K, Terada S, et al. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol (Berl) . 2008;115(5):561-575. https://doi.org/10.1007/s00401-007-0329-z 138. Munoz DG, Neumann M, Kusaka H, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol (Berl) . 2009;118(5):617-627. https://doi.org/10.1007/s00401-009-0598-9 139. Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IRA. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol (Berl) . 2009;118(5):605-616. https://doi.org/10.1007/s00401-009-0581-5 140. Lashley T, Rohrer JD, Bandopadhyay R, et al. A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brain J Neurol . 2011;134(Pt 9):2548-2564. https://doi.org/10.1093/brain/awr160 141. Bieniek KF, Josephs KA, Lin WL, Dickson DW. Neuronal intermediate filament inclusion disease may be incorrectly classified as a subtype of FTLD-FUS. Free Neuropathol . 2020;1:1-9, 9. https://doi.org/10.17879/freeneuropathology-2020-2639 142. Murakami A, Nakamura M, Nakamura Y, Kaneko S, Yakushiji Y, Kusaka H. An autopsy case report of neuronal intermediate filament inclusion disease presenting with predominantly upper motor neuron features. Neuropathol Off J Jpn Soc Neuropathol . 2021;41(5):357-365. https://doi.org/10.1111/neup.12741 143. Lee EB, Russ J, Jung H, et al. Topography of FUS pathology distinguishes late-onset BIBD from aFTLD-U. Acta Neuropathol Commun . 2013;1(9):1-11. https://doi.org/10.1186/2051-5960-1-9 144. Cairns NJ, Grossman M, Arnold SE, et al. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology . 2004;63(8):1376-1384. https://doi.org/10.1212/01.wnl.0000139809.16817.dd 145. Mackenzie IRA, Ansorge O, Strong M, et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol (Berl) . 2011;122(1):87-98. https://doi.org/10.1007/s00401-011-0838-7 146. Kobayashi Z, Tsuchiya K, Arai T, et al. Occurrence of basophilic inclusions and FUS-immunoreactive neuronal and glial inclusions in a case of familial amyotrophic lateral sclerosis. J Neurol Sci . 2010;293(1-2):6-11. https://doi.org/10.1016/j.jns.2010.03.029 147. Hewitt C, Kirby J, Highley JR, et al. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol . 2010;67(4):455-461. https://doi.org/10.1001/archneurol.2010.52
Copyright: © 2025 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |