|
|
|
Free Neuropathology 5:20 (2024) |
|
Review |
|
Neurodevelopmental disorders: 2024 update |
|
María Martínez de Lagrán1, Karen Bascón-Cardozo1, Mara Dierssen1,2,3,4 |
|
|
Corresponding author: |
|
Submitted: 22 July 2024 |
|
Keywords: Neuronal differentiation, Epigenetics, Retrotransposons, Neurogenesis, Gene regulation, Down syndrome |
|
Abstract Neurodevelopmental disorders encompass a range of conditions such as intellectual disability, autism spectrum disorder, rare genetic disorders and developmental and epileptic encephalopathies, all manifesting during childhood. Over 1,500 genes involved in various signaling pathways, including numerous transcriptional regulators, spliceosome elements, chromatin-modifying complexes and de novo variants have been recognized for their substantial role in these disorders. Along with new machine learning tools applied to neuroimaging, these discoveries facilitate genetic diagnoses, providing critical insights into neuropathological mechanisms and aiding in prognosis, and precision medicine. Also, new findings underscore the importance of understanding genetic contributions beyond protein-coding genes and emphasize the role of RNA and non-coding DNA molecules but also new players, such as transposable elements, whose dysregulation generates gene function disruption, epigenetic alteration, and genomic instability. Finally, recent developments in analyzing neuroimaging now offer the possibility of characterizing neuronal cytoarchitecture in vivo, presenting a viable alternative to traditional post-mortem studies. With a recently launched digital atlas of human fetal brain development, these new approaches will allow answering complex biological questions about fetal origins of cognitive function in childhood. In this review, we present ten fascinating topics where major progress has been made in the last year. |
|
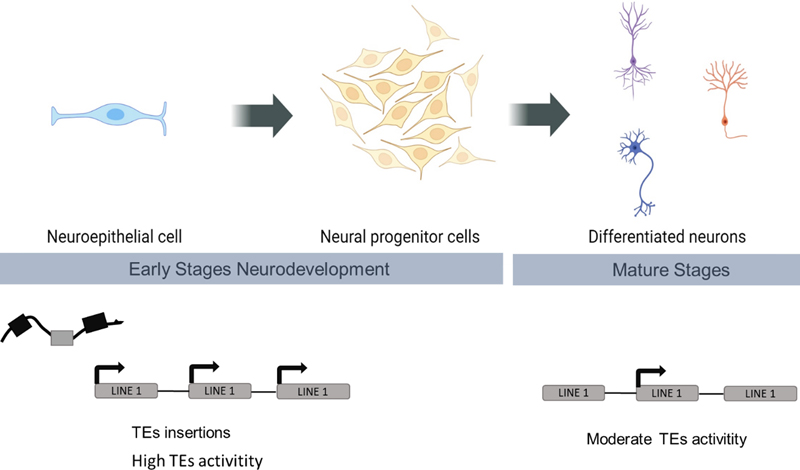
Abbreviations ABCD® - Adolescent Brain Cognitive Development℠ study; ABIDE - Autism Brain Imaging Data Exchange; ADOS - Autism Diagnostic Observation Schedule; ASD - autism spectrum disorder; BBB- blood-brain barrier; BOLD - blood-oxygen-level-dependent; CBP - CREB-binding protein; CP - cortical plate; CREBBP - CREB-binding protein; CRISPR-Cas9 - clustered regularly interspaced short palindromic repeats and CRISPR-associated protein 9; DAXX - death-associated protein; DS - Down syndrome; DTI - diffusion tensor imaging; DWI - diffusion-weighted images; ENet - elastic net; EP300 - adenovirus E1A-associated cellular p300; ER - endoplasmic reticulum; ESAM - endothelial cell adhesion molecule; fMRI - functional-MRI; GBSS - gray matter-based spatial statistics; H3K4me - histone H3 lysine 4; hCS - human cortical spheroids; hnRNPs - heterogeneous nuclear ribonucleoproteins; hPSCs - human pluripotent stem cells; HSA21 - Homo sapiens chromosome 21; ID - intellectual disability; iPSCs - induced pluripotent stem cells; ISOVF - isotropic volume fraction; JAMs - junctional adhesion molecules; KDM5C - lysine (K)-specific demethylase 5C; KDM6A - lysine (K)-specific demethylase 6A; KDM7B - lysine (K)-specific demethylase 7B; KMT2B - lysine methyltransferase 2B; LINEs‑1 - long-interspersed elements class‑1; lncRNA - long non-coding RNA; LNPK - lunapark; LTR - long-terminal repeat; MECP2 - methyl-CpG binding protein 2; ML - machine learning; MRI - magnetic resonance imaging; NDD - neurodevelopmental disorder; NDI - neurite density index; NODDI - neurite orientation dispersion and density imaging; NVU - neurovascular unit; OCLN - occludin; ODI - orientation dispersion index; oRGs - outer radial glia; oSVZ - outer subventricular zone; p300 - E1A-associated protein p300; PBMC - peripheral blood mononuclear cell; PCP preprocessed connectomes project; RBPs - RNA-binding proteins; RBFOX1 - RNA-binding Fox protein 1; ROI - regions of interest; RSI - restriction spectrum imaging; SFARI - Simons Foundation Autism Research Initiative; SHAP - SHapley Additive exPlanations; SHH - sonic hedgehog; snATACseq - single nucleus Assay for Transposase-Accessible Chromatin using sequencing; Snhg11 - small nucleolar RNA host gene 11; snRNA-seq - single nucleus RNA-sequencing; snRNPs - small nuclear RNPs; SVM - support vector machines; SVR - support vector regression; SWI/SNF - SWItch/Sucrose Non-Fermentable; TD - typically developing; TEs - transposable elements; TND - total neurite density; VZ - ventricular zone; XLMR - human X-linked mental retardation. Introduction In this new collection featuring the most relevant findings in neurodevelopmental disorders (NDDs) from 2023 and early 2024, we aimed to encompass a wide range of aspects that we believe will be of interest to neuropathologists. The selected topics range from the importance of understanding genetic and epigenetic contributions to the establishment of NDDs to the presentation of new technologies and tools for studying brain development, with a high impact on disentangling the origin of NDDs. First, we highlight genetic elements that have been historically understudied but have become in recent years more relevant in the pathogenesis of neurodegenerative disorders, new evidence suggesting their involvement in neurodevelopment. New studies suggest that de novo retrotransposition events occur during early embryogenesis and transposable elements are involved in several NDDs, such as Down syndrome (DS) (1). Although still poorly studied, we expect that their understanding of their role in the onset and evolution of NDDs will increase in the next few years. Another intriguing genetic element with a possible impact on NDDs that we spotlight in this review is the non-coding RNA, particularly the long non-coding RNA (lncRNA). LncRNAs are epigenetic regulators and thus can be determinants of the identity and functionality of postmitotic neuronal cells and the organization of the nervous system development. Around 40 % of the lncRNAs are specifically expressed in the brain, suggesting brain-specific roles and a link to neurobiological processes and NDDs. For example, the lncRNA Snhg11 is involved in DS patients with neurogenesis deficits (2,3). These findings underscore the importance of understanding genetic contributions beyond protein-coding genes. Going beyond the elements of the genome, we emphasize key regulatory mechanisms such as RNA transcription. During the pre-mRNA transcription, alternative splicing takes place to remove introns and there is increasing evidence that a spliceosome malfunction drives altered neuronal differentiation and function. In fact, a 2023 discovery shows that de novo mutations in U2AF2 and PRPF19 which encode spliceosomal subunits, are associated with intellectual disability (ID) and autism spectrum disorder (ASD) (4). A revolution in the management of big data has occurred in the last decade providing new analytical tools for disentangling complex imaging data. This is the case of the first digital atlas of human fetal brain development in which 1,059 optimal quality, three-dimensional (3D) ultrasound brain volumes from 899 fetuses have been analyzed using an automated pipeline (5). The outcome is a comprehensible tool to explore brain maturation and the origin of cognitive function in childhood. In other recent studies, new strategies to combine imaging data coming from different resources such as magnetic resonance imaging (MRI) or diffusion tensor imaging (DTI) have shed new light on the existing discrepancy in the cortical thickness across various brain regions in ASD (6,7). Finally, we discuss the use of advanced machine learning and image processing tools with different purposes such as the construction of a map showing how the fetal brain matures as pregnancy advances or the diagnosis of ASD taking into account the brain network organization (8). Another 2023 highlight is the new concept of “tightjunctionopathies”. This refers to the importance of the integrity of tight junctions in the neurovascular unit for maintaining blood-brain barrier (BBB) function and protecting the brain during development. Disruption or dysfunction of the BBB has been implicated in various NDDs, in some cases due to variants in cell adhesion proteins such as ESAM (endothelial cell-selective adhesion molecule) (9). Another relatively poorly studied aspect of NDDs is the region-specific contribution to those pathologies. The cerebellar development requires a long maturation during early childhood, becoming especially susceptible to early perturbations increasing the risk of NDDs. New findings from a single-cell genomics study at early postnatal ages indicate that early childhood inflammation prevents specific cerebellum neurons from reaching complete maturation. The authors suggest that inflammation might lead to the premature down-regulation of developmental gene expression programs with putative consequences in the onset of NDDs (10). Finally, we address the relevance of the new genes discovered through whole-exome sequencing and genome-wide association studies on brain development and NDDs, and how new in vitro experimental approaches are gaining relevance. Recent advances have combined human induced pluripotent stem cells (iPSC)-derived cerebral organoids and CRISPR-Cas9-mediated genome editing, achieving a powerful tool for investigating in vitro the role of candidate NDD genes in brain development. In this review, we have underlined two innovative studies that allow respectively to determine cell-type-specific contributions in cortical development to genetic disorders (4) and the impact of some NDD genes in the cellular migration and maturation during brain development (11). Finally, we have provided several studies using organoids and iPSc to disentangle the origin of schizophrenia by injuries during brain development (12,13). This set of studies emphasizes the importance of examining beyond neurons to understand risk genes. 1. Contribution of retrotransposition to developmental disorders Millions of transposable elements (TEs) have been accumulated throughout evolution, comprising up to 70 % of the human genome (14). Although mostly silenced via different epigenetic mechanisms, such as histone modifications and DNA methylation, the TEs activity impacts genome structure, function, and chromatin dynamics (15). TEs are classified by their mechanism of transposition into retrotransposons and DNA-transposons (16). Retrotransposons, so-called due to their "copy-and-paste" mechanism involving an RNA intermediate, are subdivided into long-terminal repeat (LTR) and non-LTR elements (17,18). One LTR subtype in particular, the Long-Interspersed Elements class‑1 (LINEs‑1), constitutes nearly 20 % of the human genome (17,18) and is the only autonomous retrotransposon encoding essential proteins for its mobility (19). LINEs‑1 are involved in diverse biological processes. During early neurodevelopment, studies have shown that active LINEs‑1 influence neural progenitor cell differentiation, contributing to neuronal diversity and brain somatic mosaicism formation (20) (Figure 1). LINEs‑1 enrichment was evidenced around genes involved in neuronal lineage commitment and function, altering gene expression levels and chromatin compaction (21). Although studies in cell cultures and animal models suggested that most de novo LINEs‑1 retrotransposition events accumulate during early embryogenesis (20), new LINEs‑1 insertions can also accumulate in adult brain tissues (22). Moreover, it was evidenced that experience-dependent LINEs‑1 retrotransposition may contribute to neuronal plasticity (23). Even though most LINEs‑1 are inactive copies unable to retrotranspose (17), they can become active. Studies have shown that during senescence, LINE‑1 elements become de-repressed and actively transpose (24), showing misregulated retrotransposition that can lead to pathological outcomes. LINEs‑1 are also dysregulated in NDDs (25,26) such as Rett’s syndrome (22), ASD, or fragile X syndrome (27)..In recent studies, altered LINEs‑1 were detected in a DS mouse model. Their altered activity was blocked by a reverse transcriptase inhibitor thus rescuing hippocampal‐dependent recognition memory (1,28). This may be related to the role of LINEs‑1 in neurogenesis which is altered in DS mouse models (3,29). In summary, despite TEs comprising more than half of the human genome, their role in brain disorders is understudied. Their pervasive presence and activity, especially LINEs‑1, seem to be essential for proper brain development and function. TEs dysregulation generates gene function disruption, epigenetic alteration, and genomic instability which may lead to NDDs. With advances in sequencing technologies and gene editing approaches, we will certainly be able to understand better the role of TEs in the future.
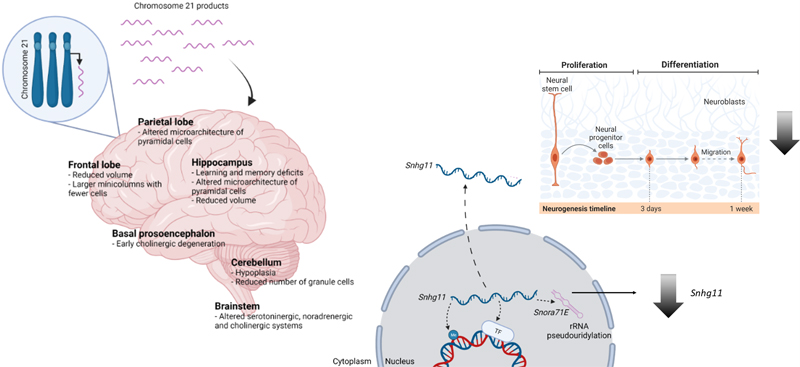
Figure 1. Transposable elements (TEs) involved in neurogenesis and cell differentiation. Scheme showing insertion and activation of LINEs‑1 at early stages in neurodevelopment. In differentiated neurons at mature stages, LINEs‑1 tend to be less active or repressed (Created with BioRender.com). 2. New epigenetic players in neurodevelopmental disorders Epigenetic mechanisms are key determinants of the identity and functionality of postmitotic neuronal cells and the organization of the nervous system development. It is thus not surprising that mutations of genes involved in chromatin regulation and genome organization lead to NDDs. This is illustrated by monogenic syndromes like the α-thalassemia mental retardation syndrome, caused by the mutation of the ATRX, a chromatin-remodeling enzyme of the SWI/SNF family that cooperates with the histone chaperone DAXX to promote nucleosome exchange (30). Histone demethylases are involved in neural development, hence mutations in KDM5C and KDM7B are found in human X-linked mental retardation (XLMR), such as the Claes-Jensen disorder (31,32). Kabuki syndrome is a mendelian disorder characterized by ID and postnatal growth deficiency that has been related to variants in genes encoding for histone demethylase KDM6A (33) and histone methyltransferase KMT2D (34). Similarly, the mutation of the X-linked gene encoding methyl-CpG binding protein 2 encoded by MECP2 results in Rett syndrome, one of the most common causes of ID in young girls (35). At the histone acetylation level, the mutation of CREBBP or EP300 transcriptional co-activator genes leads to Rubinstein-Taybi syndrome, characterized by moderate to severe ID (36). The dysfunctions manifested in these syndromes likely arise from the alteration of the epigenetic patterns required for cell fate commitment during development and neuronal maturation. In recent years, the advent of next-generation sequencing has profoundly transformed epigenetics and RNA biology with the discovery of a multitude of non-coding RNAs. Among these intriguing molecules, lncRNA are epigenetic regulators (37), acting as biological decoys, chromatin modifiers and scaffolds. Albeit lncRNAs can be found throughout the body, around 40 % are specifically expressed in the brain, suggesting brain-specific roles (38). Although these roles have only recently been investigated, multiple lncRNAs have been functionally and mechanistically linked with neurobiological processes and NDDs (39). Sierra et al. (3) showed the involvement of the lncRNA Snhg11 in DS, the most common genomic cause of ID caused by trisomy of Homo sapiens chromosome 21 (HSA21) (Figure 2). HSA21 is among the poorest chromosomes for non-coding RNAs, including microRNA-encoding genes, with the relevant exception of lncRNA genes, which are very enriched in HSA21 and are abnormally expressed in DS iPSCs (40). LncRNAs have been associated with neuronal proliferation or migration (2), but little is known about their direct mechanisms. A study proposes that lncRNAs modulate their ability to differentiate, or the migration of newly born cells to their proper location within the brain (2). Interestingly, lncRNAs might also be critical for the adult hippocampal GABAergic circuit (Liu et al., 2022), thus covering several of the DS pathogenicity mechanisms. Single-cell transcriptomics revealed that lncRNAs are abundantly expressed in individual cells and are highly cell type-specific (3,41). Using single nuclei RNA-sequencing (snRNA-seq), the authors identified Snhg11 to be specifically downregulated in the dentate gyrus of a DS mouse model and post-mortem brains of DS individuals (3). Moreover, through in vivo knockdown strategies they showed Snhg11 involvement in hippocampal-dependent cognitive phenotypes, possibly contributed by impaired adult neurogenesis. This recapitulates findings in human embryonic stem cells, in which knocking down lncRNAs blocked neurogenesis and differentiation (42). These findings underscore the importance of understanding genetic contributions beyond protein-coding genes and emphasize the role of RNA molecules and non-coding DNA in NDDs.
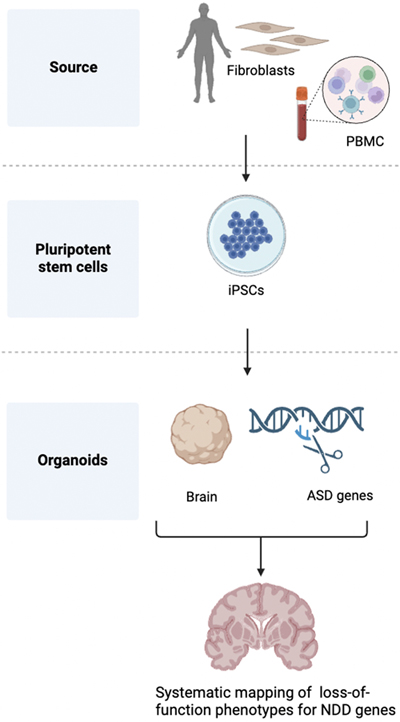
Figure 2. Snhg11, a long non-coding gene involved in Down syndrome. The left panel shows DS brain neuropathology due to the excess dosage of HSA21 products. The right panel shows the impact of a long non-coding gene (Shng11) on neurogenesis at the early stages of neurodevelopment. (Created with BioRender.com, modified from Cesar Sierra Doctoral Thesis presentation) 3. Neurodevelopmental disorders caused by spliceosome malfunction Most human protein-coding genes are transcribed as pre-mRNAs containing exons and introns, with introns removed through pre-mRNA splicing before translation. This precise process involves numerous elements and protein factors, including heterogeneous nuclear ribonucleoproteins (hnRNPs), uridine-rich small nuclear RNPs (snRNPs), the PRP19 complex, and various RNA-binding proteins (RBPs)(43). Dysregulation of these elements can significantly impact neuronal differentiation, patterning, and synaptic function, although germline variants in core spliceosome components are rarely implicated in NDDs (44). During the last year, de novo variants in spliceosome machinery have been incorporated into the list of traits related to NDDs (45–47). Li et al. (48) integrated clinical phenotyping, exome/genome sequencing, protein structure analysis, and modeling in flies and human pluripotent stem cells (hPSCs) to explore the roles of three NDD-associated genes. The authors identified 46 and 6 individuals with de novo variants in U2AF2 and PRPF19, respectively, which encode spliceosomal subunits. These variants led to neurodevelopmental phenotypes like developmental delay, ID, and ASD. Neuritogenesis was reduced in human neurons differentiated from hPSCs carrying two U2AF2 hyper-recurrent variants. Neural loss of function of the Drosophila orthologs U2af50 and Prp19 led to lethality, abnormal mushroom body patterning, and social deficits. Transcriptome profiling of deficient fly brains with CRISPR/Cas9 knock-in, and patient-derived iPSCs revealed potential splicing substrates and downstream effectors, including RNA-binding Fox protein 1 (RBFOX1). Overexpression of these effectors partially rescued neural defects in deficient flies, establishing a hierarchical genetic network of NDD-associated susceptibilities. Additionally, 6 individuals with de novo RBFOX1 missense variants exhibited similar neurodevelopmental features, highlighting a conserved genetic hotspot crucial for neurodevelopment and function. 4. First digital atlas of human fetal brain development The first digital atlas, launched in 2023 (5), illustrates the dynamics of normative maturation of each hemisphere of the fetal brain during a critical period of human development, improving the understanding of fetal brain maturation. Previous studies were limited as those were mainly carried out by a single laboratory, with usually small sample sizes, methodological heterogeneity and lack of postnatal follow-up. The normative atlas now launched is based on a prospective international cohort of healthy pregnant women, whose fetuses were accurately dated in the first trimester, with normal growth and neurodevelopment from early pregnancy to 2 years of age (49). The atlas was produced using 1,059 optimal quality, 3D ultrasound brain volumes from 899 fetuses and an automated analysis pipeline (50,51). Importantly, the study shows remarkably similar patterns of fetal brain growth and development across diverse populations, an important scientific advance in the neuroscience field. Even being much larger than previous efforts, and pooling data from the 8 geographically diverse study sites, the between-study site variability represented less than 8.0 % of the total variance of all brain measures. This result is consistent with the previously reported findings of the same INTERGROWTH-21st cohorts, for fetal skeletal growth, newborn size and infant neurocognitive development. Moreover, the atlas reproduces the findings of published MRI data (52), but with finer anatomical details in deep grey matter. The cohort used by Namburete et al. (50) included also a postnatal follow-up study to 2 years of age, so this atlas provides a unique spatiotemporal benchmark of fetal brain maturation from a large cohort with normative pre- and postnatal growth and neurodevelopment. The work revealed significant asymmetries in brain maturation since 14 gestational weeks. The most distinctive region to show asymmetry in this study was the superior frontal, which is formed by the relative overgrowth of the frontal and temporal lobes. In addition, the cerebral hemispheres follow separate maturational programs, with greater rightwards dominance in regions associated with language development without any differences between the sexes, suggestive of prioritization of language readiness. These patterns were validated in 1,487 3D brain volumes from 1,295 different fetuses. This atlas will allow answering complex biological questions about the fetal origins of cognitive function in childhood, such as how language is acquired. Using the atlas in combination with the soon-to-be-published international standards describing the complementary growth of the fetal brain will be a valuable clinical tool in specialized, referral centers when brain development appears abnormal on ultrasound. 5. Cortical microstructural imaging in neurodevelopmental disorders One of the most consistent morphological parameters for assessing abnormal brain structures in patients with ASD is changes in the cortical thickness within the frontal, temporal, parietal, and occipital lobes. These changes often correspond to specific clinical features observed in individuals with ASD (53), with core symptoms of ASD being associated with prefrontal cortex abnormalities. In fact, using cortical thickness features extracted from a small subset of brain regions as input for a classifier to identify ASD subjects, yielded accuracies of over 84.2 % when concatenating 8 regions (54). However, research in this area has yielded contradictory findings regarding the cortical thickness across various brain regions in ASD, with some reporting increases and others decreases. To complicate things even more, the spatial distribution of abnormal brain development has a high heterogeneity, even with similar pathophysiology (55). Cortical development shows an aberrant trajectory in ASD, being associated with cortical hyperplasia in early childhood, followed by a cortical plateau and subsequent decline in later stages of development (56,57). The atypical cortical developmental trajectory likely underlies structural and functional anomalies in the brain’s connectivity network (58), with cortico-cortical connectivity abnormalities not restricted to the white-matter connections but also affecting the intrinsic gray-matter architecture and connectivity within the cerebral cortex (59). Arai et al. (6) in a recent investigation focused on examining microstructural changes in gray matter among adults with ASD compared to typically developing (TD) individuals. The work employed neurite orientation dispersion and density imaging (NODDI) to measure neurite density index (NDI), orientation dispersion index (ODI), and isotropic volume fraction (ISOVF). These measures, along with DTI metrics and surface-based cortical thickness, were compared between the ASD and TD groups using voxel-wise gray matter-based spatial statistics (GBSS). Additionally, the study explored correlations between MRI-based metrics and ASD-related scores, such as the ASD-spectrum quotient, empathizing quotient, and systemizing quotient, within specific regions of interest (ROI). The analysis revealed a notable decrease in neurite density in individuals with ASD compared to the TD group, predominantly in the left prefrontal cortex regions, including the caudal middle frontal, lateral orbitofrontal, pars orbitalis, pars triangularis, rostral middle frontal, and superior frontal areas. Furthermore, there was a significant correlation between reduced empathy and lower neurite density in the left rostral middle frontal cortex, superior frontal and frontal pole areas within the ASD group. Interestingly, in another related study (7), researchers identified variable neuronal density in the cerebral cortex of individuals with ASD. This study involves a comprehensive analysis of magnetic resonance diffusion-weighted images (DWI) from the Adolescent Brain Cognitive Development℠ (ABCD®) study. They employed a restriction spectrum imaging (RSI) framework to assess total neurite density (TND). The analysis revealed a significant reduction in neurite density within the right cerebellar cortex, but an increase throughout anterior fronto-temporal and basal ganglia regions. These findings reinforce the notion that ASD patients show atypical brain growth and connectivity patterns from an early age. These recent developments in analyzing neuroimaging now offer the possibility of characterizing neuronal cytoarchitecture in vivo, presenting a viable alternative to traditional post-mortem studies. 6. Neuroimaging in the era of “Big Data” Over the past twenty years, there has been a notable increase in the generation, acquisition, and storage of data, characterizing the era known as "big data". In neuroscience, one example is the Human Brain Project, which provides different datasets, including a comprehensive, multilevel atlas of the human brain. This atlas combines data on cytoarchitecture, connectivity, chemoarchitecture, genetics, and brain function. It integrates information from both macroscopic and cellular perspectives into a unified framework and a highly detailed 3D model with microscopic resolution, distinguishing individual cortical layers and cells of the healthy brain. This significant growth in data volume, velocity, and variety along with the proliferation of machine learning (ML) toolkits has created unique opportunities for researchers. Numerous studies have introduced ML methods applied to neuroimaging data to explore various aspects of ASD. Most have focused on supervised ML algorithms, mainly based on support vector machines (SVM) to distinguish between ASD and TD. For example, using datasets sourced from the Autism Brain Imaging Data Exchange (ABIDE, analyzed with support vector regression (SVR) and elastic net (ENet)) (60,61) penalized linear regression scores on cortical thickness measurements for predicting the severity of ASD symptoms assessed with the Autism Diagnostic Observation Schedule (ADOS) (62). An international dataset of 3D ultrasound scans, previously collected using standardized methods and equipment (63), has been analyzed with advanced ML and image processing tools to construct a map showing how the fetal brain matures as pregnancy advances. The use of newly developed ML methods allowed improved visibility of brain structures in 3D ultrasound images. This results in the generation of an average representation of each cerebral hemisphere with quantification of intracranial volume variability and growth patterns between 14 and 31 gestation weeks, a critical time window of active cell proliferation, migration and synaptogenesis, as well as macroscopic changes. Despite significant advances in ML methods, several challenges remain. Many ML classification methods lack interpretability, which is particularly problematic for medical data analysis. Additionally, small datasets are prevalent, which can lead to unreliable results. Recent techniques like SHapley Additive exPlanations (SHAP) values have been developed to enhance the interpretability of ML results by identifying key features in a model. To address the issue of small datasets, data augmentation methods such as sliding windows, which segment data into smaller overlapping sections, can be employed. However, this can sometimes result in information loss that overlapping windows techniques can help mitigate. In this context, Alves et al. (8) propose a new method for diagnosing ASD that is both interpretable and suitable for small datasets. They used the preprocessed version of ABIDE, which consists of 1,112 datasets comprising 539 ASD and 573 TD with 300s BOLD (blood oxygen dependent) time series and provided by the Preprocessed Connectomes Project (PCP) dataset. Much of the recent research proposes methods based on ML but uses a single statistical parameter, ignoring brain network organization. Their approach involves classifying functional-MRI (fMRI) time series using a connectivity matrix input, which produces more accurate results compared to previous studies and is the main innovation featured by this study. They also used complex network measures to characterize brain organization differences between ASD and TD individuals and employed SHAP values for the biological interpretation of brain region connections. Additionally, they utilized a sliding window data augmentation strategy to expand the sample size, splitting time series into overlapping sections to maintain data integrity and improve model robustness. The analysis of fMRI data highlighted changes in certain brain regions associated with cognitive, emotional, learning and memory processes. The cortical networks of ASD patients displayed more segregation, less distribution of information and less connectivity compared to TD individuals. This methodology will contribute to the understanding of cerebral differences in ASD and will be useful in future to assist specialists, especially in cases involving diagnostic uncertainty. 7. Tightjunctionopathies: a new class of diseases affecting the blood-brain barrier The BBB regulates the passage of substances from the bloodstream into the brain, thereby maintaining the brain's microenvironment. It is composed of endothelial cells, astrocyte end-feet, pericytes, and a basement membrane. The BBB's integrity and functionality are crucial for normal brain development and function. Disruption or dysfunction of the BBB has been implicated in various NDDs. The BBB is a component of the neurovascular unit (NVU), a complex and dynamic interface between the brain and the blood vessels, comprising endothelial cells, pericytes, astrocytes, neurons, and the extracellular matrix. Tight junctions within the endothelial cells of the NVU are critical for maintaining the integrity of the BBB, regulating the passage of molecules and ions, and protecting the brain from potentially harmful substances. Disruption of these tight junctions is increasingly recognized as a contributing factor to NDDs (64). Tight junctions are composed of various proteins, including occludin, claudins, junctional adhesion molecules (JAMs), and cytoplasmic scaffolding proteins. These proteins interact to form a barrier that regulates paracellular permeability and maintains the homeostasis of the brain's microenvironment (65). Studies have shown changes in the expression of tight junction proteins in individuals with ASD. For instance, reduced levels of occludin (encoded by the OCLN gene, also involved in polymicrogyria), have been observed in ASD, suggesting a compromised BBB (66). The ESAM gene encodes a cell-cell attachment protein belonging to the immunoglobulin receptor family. This cell-to-cell junction is of vital importance in cells that form part of the BBB and thus, ESAM protein is essential for its integrity. Lecca et al. (9) have now identified a new rare disease in humans associated with homozygous loss-of-function variant alleles of ESAM. The study was carried out in 13 individuals including 4 fetuses, from 8 unrelated families from all over the world. The c.115del (p.Arg39Glyfs∗33) variant severely impaired the in vitro tubulogenic process of endothelial colony-forming cells, and caused a lack of ESAM expression in the capillary endothelial cells of the damaged brain. Affected individuals with bi-allelic ESAM variants presented disruption of the BBB integrity and suffered profound global developmental delay/unspecified ID, epilepsy, absent or severely delayed speech, varying degrees of spasticity, ventriculomegaly and intracranial hemorrhage/brain calcifications. Dilation of lateral ventricles, thinning of the corpus callosum, hydrocephalus, and focal white matter lesions were the most frequently observed neuroimaging abnormalities. Also, hydrocephalus was detected in all fetuses as previously reported in a patient with an ESAM homozygous nonsense variant (67). Intracranial hemorrhages, frequently associated with cerebral calcifications, were observed in all patients. The features of individuals with bi-allelic ESAM variants resemble those of other known conditions characterized by endothelial dysfunction due to mutation of genes encoding tight junction molecules, namely the JAM2 (68), JAM3 (69), and OCLN (66) genes. In summary, the integrity of tight junctions in the neurovascular unit is crucial for maintaining BBB function and protecting the brain during development. Disruption of these tight junctions can lead to increased permeability, neuroinflammation, and exposure to harmful substances, all of which contribute to the pathophysiology of various NDDs. Because of the importance of cell adhesion in the neurovascular unit, the authors propose to rename this group of diseases as “tightjunctionopathies“. 8. Zooming-in into cerebellar involvement in neurodevelopmental disorders through single-cell analysis In recent years, animal models have revealed new functions and increasing complexity of the cerebellum (70,71). However, the roles of the cerebellum in human pathology, particularly during pre- and postnatal brain development remain understudied compared to other brain regions. The human cerebellum undergoes a long maturation during early childhood being one of the first brain regions to begin developing and one of the last to reach maturity. This makes it especially susceptible to perturbations contributing to the risk of developing NDDs (72). Early-life inflammation is a clinically established risk factor for ASD and schizophrenia, but its impact on human cerebellar development is poorly understood (73). Using single-cell genomics, a study published last year found that early childhood inflammation prevents specific cerebellum neurons from reaching complete maturation (10). Ament et al. characterized the cell type-specific effects of early childhood inflammation in post-mortem cerebellar brain samples from 17 children aged one to five years, who died from inflammatory conditions, such as bacterial or viral infections or asthma. They compared them with those who died from sudden accidents but with no neurological disorder. The two groups were similar in age, gender, race/ethnicity, and time since death. The first interesting finding was that despite considerable variation among samples in the sources of inflammation, ranging from meningitis to asthma, there was a high degree of congruence in transcriptional effects across children experiencing inflammation at the time of death. The characterization of postnatal cerebellar neuronal and glial development revealed that inflammation is associated with cerebellar maturation during the postnatal period through significant, overlapping transcriptional changes in 2 types of inhibitory neurons: Purkinje and Golgi neurons. Alterations in Purkinje neuron morphology have been documented in NDDs, including ASD and schizophrenia, with the most consistent finding being a reduction in soma size (74) and a decrease in the number and density of Purkinje neurons (75). Throughout development, Purkinje neurons establish long-range synapses that connect the cerebellum to brain regions involved in cognition and emotional regulation, whereas Golgi neurons coordinate neural communication within the cerebellum. Ament et al. performed an immunohistochemical examination of post-mortem cerebellar samples that did not reveal changes in Purkinje neuron soma size but showed increased microglial activation in children who had experienced inflammation. The authors then constructed a gene regulatory network model for Purkinje neuron maturation by integrating their cell type-specific gene expression (snRNA-seq) and chromatin accessibility (single nucleus Assay for Transposase-Accessible Chromatin (snATACseq)) datasets and identified seven temporally specific gene networks in Purkinje neurons. They suggested that inflammation might lead to the premature down-regulation of developmental gene expression programs. Genes involved in NDDs such as ID, ASD, and schizophrenia (76,77) were part of the maturation- and inflammation-associated gene expression changes. Preterm birth can also lead to cerebellar underdevelopment and is often associated with chorioamnionitis with an increased risk of NDDs, such as autism. Using a preclinical experimental approach involving snRNA-seq (78) has established the role of chorioamnionitis by decreased sonic hedgehog (SHH) signaling on disrupted cerebellar maturation associated with preterm birth. Those works highlight the importance of using single-cell sequencing to zoom into the neuropathology of NDDs. 9. New cellular models are uncovering the function of neurodevelopmental disorder genes Over the past decade, whole-exome sequencing and genome-wide association studies have uncovered hundreds of ASD and other NDD genes. As promising as these discoveries are, how defects of these genes impair brain function remains largely unclear for most of those genes. This is due to the difficulties in accessing human brain tissue from NDD patients. One approach to address this question would be to investigate key features of neocortex development, using appropriate model systems and focusing on the actions of key genes that lead to NDDs. Recent advances in human iPSC-derived cerebral organoids and CRISPR-Cas9-mediated genome editing can be combined into a powerful tool for investigating molecular mechanisms involved in NDDs in in vitro systems (79). Human brain organoids recapitulate the architectural and functional features of developmental brain stages, containing apical radial glia in the ventricular zone (VZ), human-specific outer radial glia (oRGs), outer subventricular zone (oSVZ), and deep-layer and upper-layer neurons in the cortical plate (CP) (80). Several research groups have established cerebral organoids to clarify the pathogenesis of autism, Fragile X syndrome, Rett syndrome, DS, and schizophrenia (81). Last year several works offered new advanced cell models that may be instrumental in identifying the mechanisms by which candidate genes produce brain alterations. Li and collaborators (82) have developed an organoid system that couples cutting-edge technologies such as CRISPR perturbations in organoids with single-cell transcriptomic readout (CHOOSE system). Through loss-of-function assays of 19 high-risk ASD genes known to be involved in epigenetic regulation, the authors have established a developmental and cell type-specific phenotypic database. ASD genetic perturbations particularly have a strong impact on progenitors that differentiate into interneurons and oligodendrocytes and L2/3 excitatory neurons, suggesting that ASD genes are enriched in upper-layer neurons during development. The CHOOSE system opens a new frame to determine cell-type-specific contributions to genetic disorders, including NDDs (Figure 3). Going further, Pasca and colleagues (11) combined assembloids and CRISPR to identify a set of 46 genes, out from 425 associated with ASD and other NDDs, based on genetic studies of patients and the Simons Foundation Autism Research Initiative (SFARI) database. The authors had previously developed a platform to study interneuron development and migration in subpallial and forebrain assembloids (83). This 3D subdomain-specific forebrain spheroids can be assembled in vitro from human hPSCs and resemble either the dorsal or ventral forebrain and contain cortical glutamatergic or GABAergic neurons, thus being able to recapitulate the saltatory migration of GABAergic neurons from ventral to dorsal forebrain and their integration into cortical circuits observed in the fetal forebrain. Now, the authors have integrated those assembloids with CRISPR screening to investigate the involvement of NDD genes in specific aspects of interneuron development. Using this method, they discovered a group of genes that impair the generation or migration of interneurons to the CP. Notably, the loss of an endoplasmic reticulum (ER)-shaping protein, LNPK disrupts interneuron migration, indicating a previously unappreciated role of ER dynamics. The platform developed by Pasca and collaborators allows systematically map loss-of-function phenotypes for NDD genes based on stages of human interneuron development. This approach can accelerate the search for common functional deficits caused by disparate genes, enabling the grouping of neurodevelopmentally impaired patients for faster, more meaningful clinical trials and speeding treatments. However, to prevent unintended genomic alterations produced by gene editing strategies and long-term culture, the integration of advanced methods like optical genome mapping to existing protocols can provide a more thorough understanding of the genomic integrity of the in vitro cellular systems with consequences in the reliability of the research (84).
Figure 3. New cellular models are uncovering the function of neurodevelopmental disorder genes. A new organoid system that couples cutting-edge technologies such as CRISPR perturbations in organoids with single-cell transcriptomic readout (CHOOSE system) allows systematic loss-of-function assays of high-risk NDD genes (Created with BioRender.com). 10. The developmental origin of schizophrenia Schizophrenia's complex biology highlights two crucial themes: the excessive elimination of glutamatergic synapses during development and disruptions in their signaling properties. These issues may impair circuit function, contributing to the symptoms and cognitive deficits seen in schizophrenia. Since some years ago, schizophrenia has been thought to involve adverse neuropathological events beginning in early brain development (85), when disturbances in neural stem cell proliferation, neural differentiation, and synapse formation may lead to NDDs. This is the reason for using brain organoids as an interesting approach. Previous studies using iPSCs to explore neurodevelopmental processes in schizophrenia have either focused on a single stage of organoid maturation (86), or generated brain organoids from only healthy individuals. Additionally, donor variability in iPSC models can obscure case-control differences, a critical issue often overlooked (87). Moreover, schizophrenia is highly polygenic making it challenging to understand basic disease mechanisms. To preserve the disease-specific genetic context (12) decided to generate iPSC-derived organoids, called human cortical spheroids (hCS), from a genetically stratified sample of schizophrenia cases and age- and sex-matched controls. In this study, the authors conducted an extensive transcriptional profiling in human cortical spheroids derived from iPSCs by at 4 stages of hCS maturation, which enabled them to establish the persistent nature of the disturbances throughout development. The organoids grown from patients and controls differed in cell type composition but more importantly, in the expression of thousands of genes, in line with the finding that the genetic influences on schizophrenia are many and very small. However, axonal genes showed persistent dysregulation shedding light on underlying disease mechanisms. In a second study, another research group focused on the schizophrenia risk locus 15q11.2, containing 4 genes, that has over 10 % penetrance and doubles the risk for schizophrenia in individuals with unusual copy numbers in this region (13). One gene in this locus, CYFIP1, is linked to synaptic function and increases the risk for NDDs like schizophrenia and autism. Although CYFIP1 is highly expressed in microglia, the brain's immune cells, its role there remains unclear. Sheridan et al. (13) collected blood cells from healthy volunteers, isolated iPSCs, and differentiated them into microglia-like cells. Microglia are known for synaptic pruning, essential for brain development. Using CRISPR, they removed functional CYFIP1 from these cells and detected changes in microglial behavior and function that could impact microglial processes like synaptic pruning and neuronal maintenance, crucial for brain development and function. This dysfunction could contribute to CYFIP1-related neurodevelopmental and psychiatric disorders, including autism and schizophrenia. This study emphasizes the importance of examining beyond neurons to understand risk genes. Identifying risk loci is just the beginning; determining the relevant cell types and gene functions is essential for progressing from genetic associations to potential treatments. Acknowledgements The lab of MD is recognized by the Secretaria d’Universitats i Recerca del Departament d’Economia I Coneixement de la Generalitat de Catalunya (Grups consolidats 2023). We acknowledge the support of the Agencia Estatal de Investigación (PID2022-141900OB-I00). We acknowledge the support of the Spanish Ministry of Science and Innovation through the Centro de Excelencia Severo Ochoa (CEX2020-001049-S, MCIN/AEI13 /10.13039/501100011033), the Generalitat de Catalunya through the CERCA programme and to the EMBL partnership. Funding Statement The lab of M.D. acknowledges financial support from the Spanish Ministry of Science and Innovation (projects CPP2022-009659 and RTC2019-007329-1), the Spanish State Research Agency (project INTO-DS, reference PID2022-141900OB-I00), the Marato-TV3 Foundation (202212-30-31-32), and the European Comission Horizon 2020 programme (projects H2020-899986 and GO-DS21-848077). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The CRG acknowledges the support of the Spanish Ministry of Science and Innovation to the EMBL partnership, the Centro de Excelencia Severo Ochoa, and the CERCA Programme/Generalitat de Catalunya. The CIBER of Rare Diseases (CIBERER) is an initiative of ISCIII. Conflicts of Interest Statement The authors declare no conflicts of interest. All co-authors have reviewed and approved the contents of the manuscript as well as the requirements for authorship have been met. References 1. Borgognone A, Casadellà M, Martínez de Lagrán M, Paredes R, Clotet B, Dierssen M, et al. Lamivudine modulates the expression of neurological impairment-related genes and LINE-1 retrotransposons in brain tissues of a Down syndrome mouse model. Front Aging Neurosci 2024 Jul 19;16:1386944. https://www.doi.org/10.3389/fnagi.2024.1386944 2. Alammari F, Al-Hujaily EM, Alshareeda A, Albarakati N, Al-Sowayan BS. Hidden regulators: the emerging roles of lncRNAs in brain development and disease. Front Neurosci. 2024 May 22;18:1392688. https://www.doi.org/10.3389/fnins.2024.1392688 3. Sierra C, Sabariego-Navarro M, Fernández-Blanco Á, Cruciani S, Zamora-Moratalla A, Novoa EM, et al. The lncRNA Snhg11, a new candidate contributing to neurogenesis, plasticity and memory deficits in Down syndrome. Res Sq. 2023 Sep 25;rs.3.rs-3184329 4. Li D, Wang Q, Bayat A, Battig MR, Zhou Y, Bosch DGM, et al. Spliceosome malfunction causes neurodevelopmental disorders with overlapping features. J Clin Invest. 2024 an 2;134(1):e171235. https://www.doi.org/10.1172/JCI171235 5. Namburete AIL, Papież BW, Fernandes M, Wyburd MK, Hesse LS, Moser FA, et al. Normative spatiotemporal fetal brain maturation with satisfactory development at 2 years. Nature. 2023 Nov;623(7985):106–14. https://www.doi.org/10.1038/s41586-023-06568-6 6. Arai T, Kamagata K, Uchida W, Andica C, Takabayashi K, Saito Y, et al. Reduced neurite density index in the prefrontal cortex of adults with autism assessed using neurite orientation dispersion and density imaging. Front Neurol. 2023;14:1110883. https://www.doi.org/10.3389/fneur.2023.1110883 7. Christensen ZP, Freedman EG, Foxe JJ. Autism is Associated with in vivo Changes in Gray Matter Neurite Architecture. bioRxiv; 2023. p. 2023.03.25.534208 8. Alves CL, Toutain TGL de O, de Carvalho Aguiar P, Pineda AM, Roster K, Thielemann C, et al. Diagnosis of autism spectrum disorder based on functional brain networks and machine learning. Sci Rep. 2023 May 18;13(1):8072. https://www.doi.org/10.1038/s41598-023-34650-6 9. Lecca M, Pehlivan D, Suñer DH, Weiss K, Coste T, Zweier M, et al. Bi-allelic variants in the ESAM tight-junction gene cause a neurodevelopmental disorder associated with fetal intracranial hemorrhage. Am J Hum Genet. 2023 Apr 6;110(4):681–90. https://www.doi.org/10.1016/j.ajhg.2023.03.005 10. Ament SA, Cortes-Gutierrez M, Herb BR, Mocci E, Colantuoni C, McCarthy MM. A single-cell genomic atlas for maturation of the human cerebellum during early childhood. Sci Transl Med. 2023 Oct 12;15(721):eade1283. https://www.doi.org/10.1126/scitranslmed.ade1283 11. Meng X, Yao D, Imaizumi K, Chen X, Kelley KW, Reis N, et al. Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature. 2023 Oct;622(7982):359–66. https://www.doi.org/10.1038/s41586-023-06564-w 12. Akkouh IA, Ueland T, Szabo A, Hughes T, Smeland OB, Andreassen OA, et al. Longitudinal Transcriptomic Analysis of Human Cortical Spheroids Identifies Axonal Dysregulation in the Prenatal Brain as a Mediator of Genetic Risk for Schizophrenia. Biol Psychiatry. 2024 Apr 1;95(7):687–98. https://www.doi.org/10.1016/j.biopsych.2023.08.017 13. Sheridan SD, Horng JE, Yeh H, McCrea L, Wang J, Fu T, et al. Loss of Function in the Neurodevelopmental Disease and Schizophrenia-Associated Gene CYFIP1 in Human Microglia-like Cells Supports a Functional Role in Synaptic Engulfment. Biol Psychiatry. 2024 Apr 1;95(7):676–86. https://www.doi.org/10.1016/j.biopsych.2023.07.022 14. Koning APJ de, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PLOS Genet. 2011 Dec 1;7(12):e1002384. https://www.doi.org/10.1371/journal.pgen.1002384 15. Bhat A, Ghatage T, Bhan S, Lahane GP, Dhar A, Kumar R, et al. Role of Transposable Elements in Genome Stability: Implications for Health and Disease. Int J Mol Sci. 2022 Jul 15;23(14):7802. https://www.doi.org/10.3390/ijms23147802 16. Jurka J, Kohany O, Pavlicek A, Kapitonov VV, Jurka MV. Clustering, duplication and chromosomal distribution of mouse SINE retrotransposons. Cytogenet Genome Res. 2005;110(1–4):117–23. https://www.doi.org/10.1159/000084943 17. Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. https://www.doi.org/10.1146/annurev-genom-082509-141802 18. Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes. Microbiol Spectr. 2015 Apr;3(2):MDNA3-0061–2014. https://www.doi.org/10.1128/microbiolspec.MDNA3-0061-2014 19. Finnegan DJ. Retrotransposons. Curr Biol CB. 2012 Jun 5;22(11):R432-437. https://www.doi.org/10.1016/j.cub.2012.04.025 20. MacIa A, Widmann TJ, Heras SR, Ayllon V, Sanchez L, Benkaddour-Boumzaouad M, et al. Engineered LINE-1 retrotransposition in nondividing human neurons. Genome Res. 2017 Mar;27(3):335–48. https://www.doi.org/10.1101/gr.206961.116 21. Della Valle F, Thimma MP, Caiazzo M, Pulcrano S, Celii M, Adroub SA, et al. Transdifferentiation of Mouse Embryonic Fibroblasts into Dopaminergic Neurons Reactivates LINE-1 Repetitive Elements. Stem Cell Rep. 2020 Jan 2;14(1):60–74. https://www.doi.org/10.1016/j.stemcr.2019.12.002 22. Muotri AR, Marchetto MCN, Coufal NG, Oefner R, Yeo G, Nakashima K, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010 Nov 18;468(7322):443–6. https://www.doi.org/10.1038/nature09544 23. Muotri AR, Zhao C, Marchetto MCN, Gage FH. Environmental influence on L1 retrotransposons in the adult hippocampus. Hippocampus. 2009 Oct;19(10):1002–7. https://www.doi.org/10.1002/hipo.20564 24. De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019 Feb;566(7742):73–8. https://www.doi.org/10.1038/s41586-018-0784-9 25. Singer, T., et al. (2010). LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends in Neurosciences. 2010 Aug;33(8):345-54. https://www.doi.org/10.1016/j.tins.2010.04.001 26. Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011 Oct 30;479(7374):534–7. https://www.doi.org/10.1038/nature10531 27. Tan H, Qurashi A, Poidevin M, Nelson DL, Li H, Jin P. Retrotransposon activation contributes to fragile X premutation rCGG-mediated neurodegeneration. Hum Mol Genet. 2012 Jan 1;21(1):57. https://www.doi.org/10.1093/hmg/ddr437 28. Martinez de Lagran M, Elizalde-Torrent A, Paredes R, Clotet B, Dierssen M. Lamivudine, a reverse transcriptase inhibitor, rescues cognitive deficits in a mouse model of down syndrome. J Cell Mol Med. 2022 Aug;26(15):4210–5. https://www.doi.org/10.1111/jcmm.17472 29. Pons-Espinal M, Martinez de Lagran M, Dierssen M. Environmental enrichment rescues DYRK1A activity and hippocampal adult neurogenesis in TgDyrk1A. Neurobiol Dis. 2013 Dec;60:18–31. https://www.doi.org/10.1016/j.nbd.2013.08.008 30. Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010 Mar 5;140(5):678–91. https://www.doi.org/10.1016/j.cell.2010.01.003 31. Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007 Mar 23;128(6):1077–88. https://www.doi.org/10.1016/j.cell.2007.02.017 32. Jensen LR, Bartenschlager H, Rujirabanjerd S, Tzschach A, Nümann A, Janecke AR, et al. A distinctive gene expression fingerprint in mentally retarded male patients reflects disease-causing defects in the histone demethylase KDM5C. PathoGenetics. 2010 Feb 2;3:2. https://www.doi.org/10.1186/1755-8417-3-2 33. Cheon CK, Sohn YB, Ko JM, Lee YJ, Song JS, Moon JW, et al. Identification of KMT2D and KDM6A mutations by exome sequencing in Korean patients with Kabuki syndrome. J Hum Genet. 2014 Jun;59(6):321–5. https://www.doi.org/10.1038/jhg.2014.25 34. Harris J, Mahone EM, Bjornsson HT. Molecularly confirmed Kabuki (Niikawa-Kuroki) syndrome patients demonstrate a specific cognitive profile with extensive visuospatial abnormalities. J Intellect Disabil Res JIDR. 2019 Jun;63(6):489–97. https://www.doi.org/10.1111/jir.12587 35. Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005 Sep;20(9):747–53. https://www.doi.org/10.1177/08830738050200090901 36. Stevens CA. Rubinstein-Taybi Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1526/ 37. Chen J, Wang Y, Wang C, Hu JF, Li W. LncRNA Functions as a New Emerging Epigenetic Factor in Determining the Fate of Stem Cells. Front Genet. 2020 Mar 31;11. https://www.doi.org/10.3389/fgene.2020.00277 38. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012 Sep;22(9):1775–89. https://www.doi.org/10.1101/gr.132159.111 39. Aliperti V, Skonieczna J, Cerase A. Long Non-Coding RNA (lncRNA) Roles in Cell Biology, Neurodevelopment and Neurological Disorders. Non-Coding RNA. 2021 Jun 17;7(2):36. https://www.doi.org/10.3390/ncrna7020036 40. Liu SP, Fu RH, Huang YC, Chen SY, Chien YJ, Hsu CY, Tsai CH, Shyu WC, Lin SZ. Induced pluripotent stem (iPS) cell research overview. Cell Transplant. 2011;20(1):15-9. https://www.doi.org/10.3727/096368910X532828 41. Pandini C, Rey F, Cereda C, Carelli S, Gandellini P. Study of lncRNAs in Pediatric Neurological Diseases: Methods, Analysis of the State-of-Art and Possible Therapeutic Implications. Pharmaceuticals. 2023 Nov 16;16(11):1616. https://www.doi.org/10.3390/ph16111616 42. Ng SY, Johnson R, Stanton LW. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012 Feb 1;31(3):522–33. https://www.doi.org/10.1038/emboj.2011.459 43. Gebauer F, Schwarzl T, Valcárcel J, Hentze MW. RNA-binding proteins in human genetic disease. Nat Rev Genet. 2021 Mar;22(3):185–98. https://www.doi.org/10.1038/s41576-020-00302-y 44. Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016 Jan;17(1):19–32. https://www.doi.org/10.1038/nrg.2015.3 45. Kittock CM, Saifeddine M, Straight L, Ward DI. U2AF2 variant in a patient with developmental delay, dysmorphic features, and epilepsy. Am J Med Genet A. 2023 Jul;191(7):1968–72. https://www.doi.org/10.1002/ajmg.a.63041 46. Wang X, You B, Yin F, Chen C, He H, Liu F, et al. A presumed missense variant in the U2AF2 gene causes exon skipping in neurodevelopmental diseases. J Hum Genet. 2023 Jun;68(6):375–82. https://www.doi.org/10.1038/s10038-023-01141-5 47. Engal E, Oja KT, Maroofian R, Geminder O, Le TL, Mor E, et al. Biallelic loss of function variants in WBP4, encoding a spliceosome protein, result in a variable neurodevelopmental delay syndrome. MedRxiv Prepr Serv Health Sci. 2023 Jun 27;2023.06.19.23291425 48. Li D, Wang Q, Bayat A, Battig MR, Zhou Y, Bosch DGM, et al. Spliceosome malfunction causes neurodevelopmental disorders with overlapping features. J Clin Invest. 2024 Jan 20;134(1). https://www.doi.org/10.1172/JCI171235 49. Villar J, Cheikh Ismail L, Staines Urias E, Giuliani F, Ohuma EO, Victora CG, et al. The satisfactory growth and development at 2 years of age of the INTERGROWTH-21st Fetal Growth Standards cohort support its appropriateness for constructing international standards. Am J Obstet Gynecol. 2018 Feb;218(2S):S841-S854.e2. https://www.doi.org/10.1016/j.ajog.2017.11.564 50. Namburete AIL, Xie W, Yaqub M, Zisserman A, Noble JA. Fully-automated alignment of 3D fetal brain ultrasound to a canonical reference space using multi-task learning. Med Image Anal. 2018 May;46:1–14. https://www.doi.org/10.1016/j.media.2018.02.006 51. Moser, F., Huang, R., Papiez, B. W. & Namburete, A. I. L. BEAN: brain extraction and alignment network for 3D fetal neurosonography. NeuroImage. 2022 Sep 1;258, 119341. https://www.doi.org/10.1016/j.neuroimage.2022.119341 52. Gholipour A, Rollins CK, Velasco-Annis C, Ouaalam A, Akhondi-Asl A, Afacan O, et al. A normative spatiotemporal MRI atlas of the fetal brain for automatic segmentation and analysis of early brain growth. Sci Rep. 2017 Mar 28;7(1):476. https://www.doi.org/10.1038/s41598-017-00525-w 53. Doyle-Thomas KAR, Duerden EG, Taylor MJ, Lerch JP, Soorya LV, Wang AT, et al. Effects of age and symptomatology on cortical thickness in autism spectrum disorders. Res Autism Spectr Disord. 2013 Jan;7(1):141–50. https://www.doi.org/10.1016/j.rasd.2012.08.004 54. Squarcina L, Nosari G, Marin R, Castellani U, Bellani M, Bonivento C, et al. Automatic classification of autism spectrum disorder in children using cortical thickness and support vector machine. Brain Behav. 2021 Jul 15;11(8):e2238. https://www.doi.org/10.1002/brb3.2238 55. Zabihi M, Oldehinkel M, Wolfers T, Frouin V, Goyard D, Loth E, et al. Dissecting the Heterogeneous Cortical Anatomy of Autism Spectrum Disorder Using Normative Models. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019 Jun;4(6):567–78. https://www.doi.org/10.1016/j.bpsc.2018.11.013 56. Zielinski BA, Prigge MBD, Nielsen JA, Froehlich AL, Abildskov TJ, Anderson JS, et al. Longitudinal changes in cortical thickness in autism and typical development. Brain J Neurol. 2014 Jun;137(Pt 6):1799–812. https://www.doi.org/10.1093/brain/awu083 57. Bieneck V, Bletsch A, Mann C, Schäfer T, Seelemeyer H, Herøy N, et al. Longitudinal Changes in Cortical Thickness in Adolescents with Autism Spectrum Disorder and Their Association with Restricted and Repetitive Behaviors. Genes. 2021 Dec 20;12(12):2024. https://www.doi.org/10.3390/genes12122024 58. Wass S. Distortions and disconnections: disrupted brain connectivity in autism. Brain Cogn. 2011 Feb;75(1):18–28. https://www.doi.org/10.1016/j.bandc.2010.10.005 59. Ecker C, Ronan L, Feng Y, Daly E, Murphy C, Ginestet CE, et al. Intrinsic gray-matter connectivity of the brain in adults with autism spectrum disorder. Proc Natl Acad Sci. 2013 Aug 6;110(32):13222–7. https://www.doi.org/10.1073/pnas.1221880110 60. Di Martino A, Yan CG, Li Q, Denio E, Castellanos FX, Alaerts K, et al. The autism brain imaging data exchange: towards a large-scale evaluation of the intrinsic brain architecture in autism. Mol Psychiatry. 2014 Jun;19(6):659–67. https://www.doi.org/10.1038/mp.2013.78 61. Moradi E, Khundrakpam B, Lewis JD, Evans AC, Tohka J. Predicting symptom severity in autism spectrum disorder based on cortical thickness measures in agglomerative data. NeuroImage. 2017 Jan 1;144(Pt A):128–41. https://www.doi.org/10.1016/j.neuroimage.2016.09.049 62. Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC, et al. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000 Jun;30(3):205–23. https://www.doi.org/10.1023/A:1005592401947 63. Hesse LS, Aliasi M, Moser F, INTERGROWTH-21(st) Consortium, Haak MC, Xie W, et al. Subcortical segmentation of the fetal brain in 3D ultrasound using deep learning. NeuroImage. 2022 Jul 1;254:119117. https://www.doi.org/10.1016/j.neuroimage.2022.119117 64. Moretti R, Pansiot J, Bettati D, Strazielle N, Ghersi-Egea JF, Damante G, et al. Blood-brain barrier dysfunction in disorders of the developing brain. Front Neuroscience. 2015 Feb 17; 9:40. https://www.doi.org/10.3389/fnins.2015.00040 65. Virgintino D, Errede M, Robertson D, Capobianco C, Girolamo F, Vimercati A, et al. Immunolocalization of tight junction proteins in the adult and developing human brain. Histochem Cell Biol. 2004 Jul;122(1):51–9. https://www.doi.org/10.1007/s00418-004-0665-1 66. O’Driscoll MC, Daly SB, Urquhart JE, Black GCM, Pilz DT, Brockmann K, et al. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am J Hum Genet. 2010 Sep 1;87(3):354–64. https://www.doi.org/10.1016/j.ajhg.2010.07.012 67. Jin SC, Dong W, Kundishora AJ, Panchagnula S, Moreno-De-Luca A, Furey CG, et al. Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus. Nat Med. 2020 Nov;26(11):1754–65. https://www.doi.org/10.1038/s41591-020-1090-2 68. Cen Z, Chen Y, Chen S, Wang H, Yang D, Zhang H, et al. Biallelic loss-of-function mutations in JAM2 cause primary familial brain calcification. Brain J Neurol. 2020 Feb 1;143(2):491–502. https://www.doi.org/10.1093/brain/awz392 69. Mochida GH, Ganesh VS, Felie JM, Gleason D, Hill RS, Clapham KR, et al. A Homozygous Mutation in the Tight-Junction Protein JAM3 Causes Hemorrhagic Destruction of the Brain, Subependymal Calcification, and Congenital Cataracts. Am J Hum Genet. 2010 Dec 10;87(6):882–9. https://www.doi.org/10.1016/j.ajhg.2010.10.026 70. Van Overwalle F, Manto M, Cattaneo Z, Clausi S, Ferrari C, Gabrieli JDE, et al. Consensus Paper: Cerebellum and Social Cognition. Cerebellum Lond Engl. 2020;19(6):833–68. https://www.doi.org/10.1007/s12311-020-01155-1 71. De Zeeuw CI, Lisberger SG, Raymond JL. Diversity and dynamism in the cerebellum. Nat Neurosci. 2021 Feb;24(2):160–7. https://www.doi.org/10.1038/s41593-020-00754-9 72. Sathyanesan A, Zhou J, Scafidi J, Heck DH, Sillitoe RV, Gallo V. Emerging connections between cerebellar development, behaviour and complex brain disorders. Nat Rev Neurosci. 2019 May;20(5):298–313. https://www.doi.org/10.1038/s41583-019-0152-2 73. Jiang NM, Cowan M, Moonah SN, Petri WA. The Impact of Systemic Inflammation on Neurodevelopment. Trends Mol Med. 2018 Sep;24(9):794–804. https://www.doi.org/1 0.1016/j.molmed.2018.06.008 74. Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, et al. Consensus Paper: Pathological Role of the Cerebellum in Autism. Cerebellum Lond Engl. 2012 Sep;11(3):777–807. https://www.doi.org/10.1007/s12311-012-0355-9 75. Wegiel J, Flory M, Kuchna I, Nowicki K, Ma SY, Imaki H, et al. Stereological study of the neuronal number and volume of 38 brain subdivisions of subjects diagnosed with autism reveals significant alterations restricted to the striatum, amygdala and cerebellum. Acta Neuropathol Commun. 2014 Sep 18;2:141. https://www.doi.org/10.1186/s40478-014-0141-7 76. Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet Lond Engl. 2015 Apr 4;385(9975):1305–14. https://www.doi.org/10.1016/S0140-6736(14)61705-0 77. Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism. 2013 Oct 3;4(1):36. https://www.doi.org/10.1186/2040-2392-4-36 78. Newman J, Tong X, Tan A, Yeasky T, De Paiva VN, Presicce P, et al. Chorioamnionitis accelerates granule cell and oligodendrocyte maturation in the cerebellum of preterm nonhuman primates. J Neuroinflammation. 2024 Jan 10;21(1):16. https://www.doi.org/10.1186/s12974-024-03012-y 79. Kelava I, Lancaster MA. Stem Cell Models of Human Brain Development. Cell Stem Cell. 2016 Jun 2;18(6):736–48. https://www.doi.org/10.1016/j.stem.2016.05.022 80. Kelley KW, Pașca SP. Human brain organogenesis: Toward a cellular understanding of development and disease. Cell. 2022 Jan 6;185(1):42–61. https://www.doi.org/1 0.1016/j.cell.2021.12.031 81. Qian X, Su Y, Adam CD, Deutschmann AU, Pather SR, Goldberg EM, et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell. 2020 May 7;26(5):766-781.e9. https://www.doi.org/10.1016/j.stem.2020.02.002 82. Li C, Fleck JS, Martins-Costa C, Burkard TR, Themann J, Stuempflen M, et al. Single-cell brain organoid screening identifies developmental defects in autism. Nature. 2023 Sep;621(7978):373–80. https://www.doi.org/10.1038/s41586-023-06473-y 83. Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017 May 4;545(7652):54–9. https://www.doi.org/10.1038/nature22330 84. Gallego Villarejo L, Gerding WM, Bachmann L, Hardt LHI, Bormann S, Nguyen HP, et al. Optical Genome Mapping Reveals Genomic Alterations upon Gene Editing in hiPSCs: Implications for Neural Tissue Differentiation and Brain Organoid Research. Cells. 2024 Jan;13(6):507. https://www.doi.org/10.3390/cells13060507 85. Owen MJ, O’Donovan MC, Thapar A, Craddock N. Neurodevelopmental hypothesis of schizophrenia. Br J Psychiatry J Ment Sci. 2011 Mar;198(3):173–5. https://www.doi.org/10.1192/bjp.bp.110.084384 86. Notaras M, Lodhi A, Dündar F, Collier P, Sayles NM, Tilgner H, et al. Schizophrenia is defined by cell-specific neuropathology and multiple neurodevelopmental mechanisms in patient-derived cerebral organoids. Mol Psychiatry. 2022 Mar;27(3):1416–34. https://www.doi.org/10.1038/s41380-021-01316-6 87. Hoffman GE, Schrode N, Flaherty E, Brennand KJ. New considerations for hiPSC-based models of neuropsychiatric disorders. Mol Psychiatry. 2019 Jan;24(1):49–66. https://www.doi.org/10.1038/s41380-018-0029-1
Copyright: © 2024 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |