|
|
|||||||||||||||||||||||
|
Free Neuropathology 5:16 (2024) |
|||||||||||||||||||||||
|
Original Paper |
|||||||||||||||||||||||
|
Fast-track neuropathological screening for neurodegenerative diseases |
|||||||||||||||||||||||
|
Benjamin Englert1,2,*, Sigrun Roeber1, Thomas Arzberger1,3, Viktoria Ruf1, Otto Windl1, Jochen Herms1,2,4 |
|||||||||||||||||||||||
* Present address: Department of Pathology, University of Helsinki and HUS Diagnostic Center, Helsinki University Hospital, Helsinki, Finland |
|||||||||||||||||||||||
|
Corresponding author: |
|||||||||||||||||||||||
|
Submitted: 07 June 2024 |
|||||||||||||||||||||||
|
Keywords: Neuropathology, Dementia, Movement disorders, Prion disease, Alzheimer disease, Lewy body disease |
|||||||||||||||||||||||
|
Abstract
Background: The postmortem diagnostic of individuals having suffered presumptive neurodegenerative disease comprises exclusion of a prion disease, extensive brain sampling and histopathological evaluation, which are resource-intensive and time consuming. To exclude prion disease and to achieve prompt accurate preliminary diagnosis, we developed a fast-track procedure for the histopathological assessment of brains from patients with suspected neurodegenerative disease. |
|||||||||||||||||||||||
|
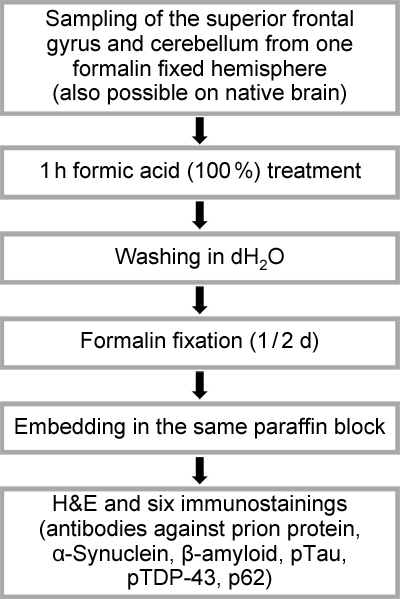
Introduction Dementia or cognitive impairment are common clinical diagnoses; however, they often lack neuropathological verification by autopsy. Compared to other underlying causes of death, dementia lowers the chances of an autopsy to be performed [1]. The low autopsy rates, in turn, lead to a loss of expertise for diagnosing neurodegenerative diseases with high precision and according to well-established standards. A study by Selvackadunko et al., comparing the antemortem clinical diagnosis and postmortem neuropathological diagnosis revealed that in one third of cases the in-life and the neuropathological diagnosis differed [2]. A Swedish study found a full accordance of clinical and neuropathological dementia diagnosis in 49 % of cases, in a further 14 % the clinical diagnosis corresponded with some but not all the diagnoses after postmortem evaluation [3]. In Alzheimer’s disease (AD), the commonest neurodegenerative disease (NDD), a study showed that 119 out of 533 clinically diagnosed AD cases did not fulfil the neuropathological criteria for definite AD at autopsy, with dementia with Lewy bodies (DLB), vascular dementia, frontotemporal lobar degeneration (FTLD) and hippocampal sclerosis being the most frequently encountered AD mimics [4]. In progressive supranuclear palsy (PSP), only 78 % of clinically diagnosed cases were histologically confirmed as indeed being a PSP, clinically incorrect classified cases histologically were classified as Parkinson’s disease (PD), AD, multiple system atrophy (MSA), Pick’s disease (PiD), motor neuron disease (MND) and corticobasal degeneration (CBD) [5]. Furthermore, prion disease can be mistaken for various other clinical entities, especially in the elderly due to a less consistent clinical phenotype and a high incidence of dementia [6]. Even though new developments in tau positron emission tomography imaging and cerebrospinal fluid analysis have very much improved the clinical diagnosis of NDDs in the last three years, these expensive methods are often not available to clinicians [7]. The conventional neuropathological workup for brains of patients with suspected neurodegenerative diseases is extensive, since it typically includes a comprehensive and detailed characterization of all pathologies and co-pathologies present, particularly in a brain bank setting. For this, the analysis of at least seven or eight tissue blocks from different brain regions is required. A report by Kovacs and Budka from the Vienna brain bank describes five immunostainings on seven brain regions as minimal number to assess protein deposition in NDD [8]. The National Institute of Ageing guidelines recommend the sampling of at least eight brain regions for AD, six for Lewy body disease (LBD) [9]. Since up to five immunohistological stainings per region are necessary, these approaches lead to about 50 stained sections to be analysed under the microscope, which is costly and time-consuming, taking a considerable time until a final report can be provided. In contrast, the priority for clinicians and relatives is to receive information about the definite (main) diagnosis in a timely manner. To reduce costs and effort, condensed protocols for common neurodegenerative alterations have been deployed, essentially based on the protocol by Flanagan et al. [10]. This protocol requires sampling of 20 brain regions (sometimes bilateral sampling of the same region is needed), with four regions embedded in the same tissue cassette, giving a total of five paraffin blocks [10]. Following a nontiered approach, all blocks are stained with HE and Luxol fast blue, and each block with an additional specific stain (Bielschowsky silver impregnation or immunohistochemical stains for α-Synuclein, β-Amyloid or phospho-Tau) [10]. Clement et al. further simplified the protocol by unilateral sampling of 12 brain regions embedded in six tissue cassettes, with better preservation of neuroanatomical relationships [1]. Amended protocols have been published for the diagnosis of frontotemporal lobar degeneration (FTLD) and limbic-predominant age-related TDP-43 encephalopathy neuropathological changes (LATE-NC) [12,13]. Condensed protocols have been reliably applied in clinical and medicolegal autopsy settings [14,15,16]. Previous studies have already addressed the question of the utility of small-sized tissue samples of few selected brain regions for NDD diagnostics. Venetti et al. performed simulated brain biopsies on 73 autopsy cases of various NDD and compared the outcome between assessments of the mid-frontal cortex alone or four brain regions (frontal, temporal, parietal cortex and basal ganglia) to the final autoptic diagnoses [17]. Considering solely the frontal sample, the authors indicate an average sensitivity of 64 % and a specificity of 43 %, and 92 % sensitivity and 71 % specificity for all four brain regions, with the highest sensitivities found for FTLD with TDP-43 inclusions (FTLD-TDP, 88 %) and AD (80 %) and the lowest for PSP (0 %) [17]. A study by King et al. evaluated 62 NDD cases using 1 cm3 of prefrontal and middle temporal cortex and compared the results with gold standard autopsy diagnoses [18]. The sensitivity of assessing the frontal lobe alone was 81 % and the specificity 79 % [18]. These figures increased to a sensitivity of 85 % and a specificity of 83 % when taking both regions into account [18]. The diagnostic accuracy of evaluating frontal lobe specimens was high e.g. in AD, CBD and MSA (100 % of correct diagnoses) and low in PSP (0 %), ascribed by the authors to sparse expression of tau-positive PSP lesions in the neocortex [18]. We use the below-described protocol for screening not only to obtain a rather quick working diagnosis that allows to diagnose more than 96 % of NDDs but also to protect laboratory personnel from extensive contact with potentially prion-infected tissue. Material and methods In our study we included 133 cases who underwent neuropathological assessment as brain donors to the Neurobiobank Munich (NBM), located at the Centre for Neuropathology and Prion research, Ludwig-Maximilians-University (LMU), Munich, Germany. The collection of tissue for the NBM was approved by the ethics committee of the medical faculty of the LMU (No. 345‑13). After the brain had been removed, one hemisphere was cryopreserved while the other hemisphere was fixed for at least two weeks in 4 % buffered formalin. After sufficient fixation, small tissue specimens (1 x 1 x 0.5 cm) from the superior frontal gyrus (approximately BA 8) and the cerebellum (lateral cerebellar cortex) were sampled, treated with 100 % formic acid for one hour [19], rinsed with water, fixed in formalin for half a day and embedded into the same paraffin block. Tissue sections were cut and stained with H&E and subjected to six immunohistochemical stainings for prion protein (clone L42, targeting an epitope on the first alpha helix [20], 1 : 50 - 1 : 100, pre-treatment with proteinase K, own), α-Synuclein (clone 42, 1 : 2000, BD Transduction Laboratories); β-Amyloid (clone 4G8, 1 : 2000, Covance); phospho-Tau (clone AT8, 1 : 200, ThermoFisher Scientific); phospho-TDP-43 (clone 1D3, 1 : 50, own); p62 (clone 3/P62 lck ligand, 1 : 100, BD Transduction Laboratories)). Stainings were performed using an automatic staining system (Roche, Ventana BenchMark Ultra). If required and in accordance with the respective clinical diagnosis, various other immunostainings (e.g. FUS, 3-repeat tau) or silver impregnation techniques (Bielschowsky, Gallyas) were occasionally performed. A schematic overview of the work-up is presented in figure 1. Firstly, we assessed the H&E stain for the presence of general neurodegenerative features (atrophy, neuronal loss, spongiosis, gliosis) and various other pathological alterations such as tissue ischemia/hypoxia, haemorrhage, inflammation or neoplasms as well as vascular pathologies. Furthermore, disease specific alterations e.g. senile plaques in AD or ballooned neurons in CBD were examined on an H&E stain [21,22].
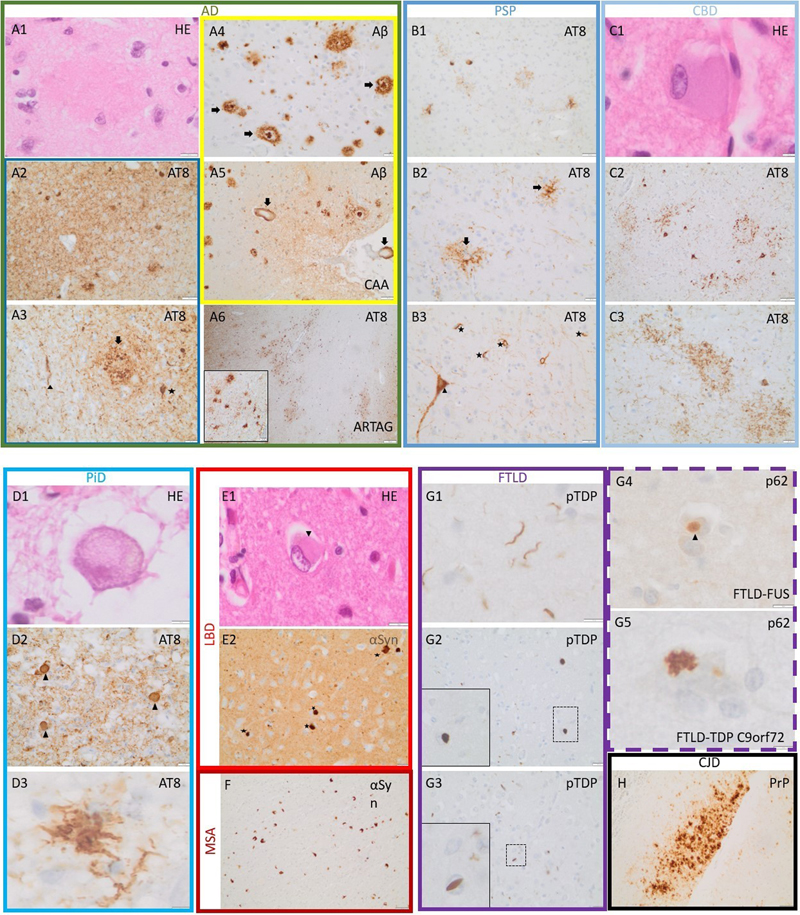
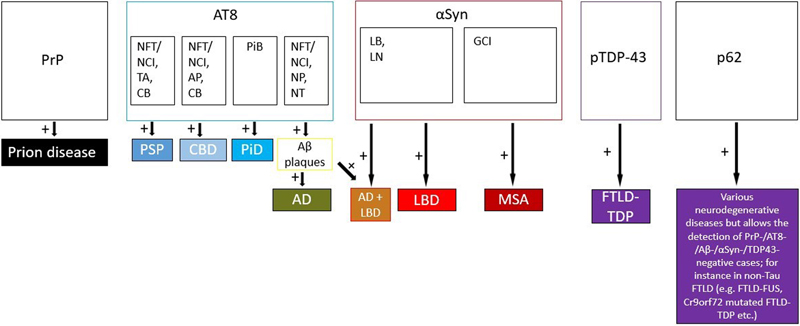
Figure 1: Process flow of fast-track screening histology For further neuropathological diagnosis, we evaluated the presence of the following morphological features by immunohistochemistry. Prion protein immunoreactive structures in a synaptic, vacuolar, laminar cortical or coarse pattern, plaque-like focal deposits or amyloid-kuru plaques were searched to diagnose prion disease [23]. 4G8-positive Amyloid β (Aβ) plaques, tau-immunoreactive neurofibrillary tangles (NFT), neuropil threads (NT) and neuritic plaques (NP) were used for the fast-track diagnosis of higher stages of AD associated pathology [9,24]. The diagnosis of PSP was made when NT, NFT, neuronal cytoplasmic inclusions (NCI), tufted astrocytes (TA) and oligodendroglial coiled bodies (CB) were visible [25,26,27]. NCI, NT, CB and most notably astrocytic plaques (AP) were highly suggestive for CBD [27,28]. Pick bodies (PiB) were the characteristic hallmark of PiD [29]. Intraneuronal α-Synuclein positive Lewy bodies (LB), extracellular Lewy body-like inclusions, Lewy neurites (LN) and grain-like cytoplasmic inclusions are present in LBD [30]. Given the overlapping and, if at all, not readily distinguishable neuropathological features of PD, DLB and Parkinson’s disease dementia [31,32], we summarized these three entities under the umbrella term LBD. MSA is characterised by α-Synuclein immunoreactive glial cytoplasmic inclusions (GCI) [3]. Phospho-TDP-43 positive neuronal and glial intracytoplasmic inclusions as well as dystrophic neurites can be found in FTLD-TDP [34]. For FTLD cases without Tau- or TDP-43-positive inclusions, immunostaining for the ubiquitin binding protein p62 detecting cytoplasmic glial/neuronal inclusions, neuronal nuclear inclusions, threads provides a valuable tool prompting further immunohistochemical characterisation [34,35]. Representative micrographs of characteristic histomorphological findings and protein deposits are shown in figure 2. A decision diagram showing a representative guide to make a screening diagnosis according to characteristic histological findings is shown in figure 3.
Figure 2: Representative micrographs of characteristic histological findings. A1: Eosinophilic plaque in AD. A2: Strongly stained neuropil (AT8) with numerous intraneuronal inclusions AD. A3: AT8-positive neuronal inclusion (star), neuropil thread (arrowhead) and a neuritic plaque (arrow) in AD. A4: Cored (arrows) and diffuse Aβ-plaques in AD. A5: Parenchymal and leptomeningeal amyloid deposits in vessel walls (cerebral amyloid angiopathy, arrows) in an AD patient. A6: Signs of aging-related tau astrogliopathy with perivascular accentuation (inset with higher magnification) as common co-pathology in AD. B1, B2, B3: PSP with less pronounced positivity of the neuropil, frequent TA (arrows), NFT (arrowhead) and oligodendroglial CB (stars). C1: Ballooned neuron in CBD. C2, C3: Overview of a CBD case showing moderate NT, neuronal inclusions and AP (C3). Hallmarks of PiD include a ballooned neuron (D1), AT8-positive Pick bodies (D2, arrowheads) as well as a ramified astrocyte (D3). E1, E2: Lewy bodies in LDB (arrowhead, stars; rarely visible in HE-stain). F: numerous cerebellar GCI in MSA. G1, G2, G3: Various pTDP-43-positive dystrophic neurites (G1), compact neuronal (G2) and neuronal cat-eye shaped (G3) inclusions in FTLD-TDP. G4: TDP-43-negative, p62-positive globular inclusion in FTLD-FUS (arrowhead). G5: Typical star-shaped p62-positive inclusion in FTLD-TDP with C9orf72 mutation. H: Deposits of prion protein in Creutzfeldt-Jakob disease. Images show stainings with HE and with antibodies against β-Amyloid (Aβ), phospho-tau (AT8), α-Synuclein (αSyn), phospho-TDP43 (pTDP), p62 and prion protein (PrP). Scale bars: C1, D1, D3, G5: 5 μm; A1, E1, G1, G4: 10 μm; A3, A4 A6 (inset), B2, B3, C3, D2, E2, G2, G3: 20 μm; A2, A5, B1, C2, F, H: 50 μm; A6: 200 μm.
Figure 3: Decision diagram guiding the investigator to establish a screening diagnosis based on characteristic histological findings. PrP: prion protein, AT8: phospho-Tau, NFT: neurofibrillary tangles, NCI: neuronal cytoplasmic inclusions, TA: tufted astrocytes, CB: coiled bodies, AP: astrocytic plaques, PiB: Pick bodies, NP: neuritic plaques, NT: neuropil threads, Aβ: β-Amyloid, αSyn: α-Synuclein, LB: Lewy bodies, LN: Lewy neurites, GCI: glial cytoplasmic inclusions, PSP: progressive supranuclear palsy, CBD: corticobasal degeneration, PiD: Pick’s disease, AD: Alzheimer’s disease, LBD: Lewy body disease, MSA: multiple system atrophy, FTLD: frontotemporal lobar degeneration. A limited neuropathological disease staging was also performed. A Thal-Score for Aβ deposits was assigned to a stage of either (at least) 1 (frontal beta amyloid plaques) or 5 (additional Aβ plaques detected in the cerebellum) [36]. The presence of frontal tau-immunoreactive neurofibrillary pathology allowed for assigning a Braak and Braak stage of at least IV [37]. Recognition and staging of additional CAA according to Thal et al. [38] is limited to stages 1 (if frontal pathology is present) or (at least) 2 (with cerebellar amyloid angiopathy). Frontal α-Synuclein inclusions lead to designation of a Braak stage of 6 [39]. A full histological work-up according to a standardized, well-established NBM protocol was performed on formalin-fixed hemispheres after complete fixation, taking both the working diagnosis obtained by screening histology and the deceased's medical history into account for a more targeted evaluation. The final neuropathological diagnoses were made in accordance with established guidelines for the histological diagnosis of NDD [9,24,25,30,33,40,41,42]. Point estimates and 95 % confidence intervals (CI) for sensitivity and specificity comparing fast-track to final diagnoses were calculated for each disease group using GraphPad Prism version 9.4.1, GraphPad Software, San Diego, California, USA. Statistical significance was set at the 0.05 level (Fisher’s exact test). Kappa coefficients for inter-rater variability between fast-track and final diagnoses were calculated with a GraphPad online tool (https://www.graphpad.com/quickcalcs/kappa1/). According to the Kappa values the agreement was considered as poor (kappa = 0), slight (0 - 0.2), fair (0.21 - 0.4), moderate (0.41 - 0.6) substantial (0.61 - 0.8) or almost perfect (0.81 - 1.0) [43]. Results Fast-track histology was analysed in 133 cases (58 females, 75 males, median age at death: 71.1 years; age range: 38.7 - 95.1 years) which were enrolled in a brain donation program for brain banking and thus underwent brain autopsy at NBM between March 2015 and December 2020. From 156 cases initially selected, we excluded 15 cases because of suspected motor neuron disease (MND)/amyotrophic lateral sclerosis (since spinal samples were not assessed on screening histology), three cases with clinical restless legs syndrome, one case with suspected Fahr’s disease and four cases with Huntington’s disease already genetically confirmed during lifetime. The main histological NDD diagnoses without consideration of additional pathology besides LBD pathology in AD shown in table 1 were: PSP (n = 26, 19.5 %), AD without LBD pathology (n = 24, 18.05 %), LBD (n = 22, 16.5 %), AD with LBD (n = 13, 9.77 %), CBD (n = 14, 10.5 %), MSA (n = 14, 10.5 %) non-tau FTLD (FTLD-TDP, FTLD-FUS; n = 9, 6.7 %), prion disease (n = 4, 3 %), PiD (n = 3, 2.3 %). In four cases (3 %), no diagnosis could be made using fast-track histology and additional histological work-up was hence required for neuropathological diagnosis. Those cases were later classified as PSP (two cases), FTLD non-tau and LBD (one case each). In 80 cases (60.2 %), the suspected clinical diagnosis could be confirmed by fast-track histology, whereas in 49 (39.8 %) the clinical diagnosis was either ambiguous or different from the histopathological diagnosis. When comparing the initial fast-track diagnosis to the diagnosis made after subsequent conventional pathological work-up of the brain, it became evident that in 128 cases (96.2 %) the NDD entity determined by fast-track diagnosis was correct and only co-pathologies or correct disease staging needed to be determined. The fast-track diagnosis needed revision in only one case of an unusual tauopathy initially classified as PSP (3 %). In four cases, the analysis of additional brain regions after fast-track histology was required to reach a conclusive histopathological diagnosis, as mentioned above. There was a high sensitivity for the diagnosis, considering only the main pathology without additional pathology, for AD, Braak and Braak stage 5 or 6 (1.0, CI = 0.9059 - 1.00, p < 0.0001), CBD (1.0, CI = 0.7847 - 1.00, p < 0.0001), LBD (1.0, CI = 0.8513 - 1.00, p < 0.0001), PiD (1.0, CI = 0.4385 - 1.00, p < 0.0001), MSA (1.0, CI = 0.7847 - 1.00, p < 0.0001) and FTLD non-tau (1.0, CI = 0.7009 - 1.00, p < 0.0001). The sensitivity for the diagnosis of PSP was 0.96 (CI = 0.8046 - 0.9979, p < 0.0001). The respective specificity was for AD 1.0 (CI = 0.9615 - 0, p < 0.0001), for CBD 1.0 (CI = 0.9687 - 1.0, p < 0.0001), for LBD 0.9910 (CI = 0.9507 - 0.9995, p < 0.0001), for PiD 1.0 (CI = 0.9713 - 1.0, p < 0.0001), for MSA 1.0 (CI = 0.9687 - 1.0, p < 0.0001), for FTLD non-tau 0.9919 (CI = 0.9557 - 0.9996, p < 0.0001) and for PSP 0.9815 (CI = 0.9350 - 0.9967, p < 0.0001).
The Kappa coefficient for inter-rater variability comparing fast-track and final histology was as follows: AD 1.0 (CI = 1.0 - 1.0), CBD 1.0 (CI = 1.0 - 1.0), PiD 1.0 (CI = 1.0 - 1.0), MSA 1.0 (CI = 1.0 - 1.0), FTLD non-tau 0.943 (CI = 0.833 - 1.0), LBD 0.973 (CI = 0.921 - 1.0), PSP 0.927 (CI = 0.846 - 1.0). Among the cases referred to our brain bank with various clinical diagnoses such as atypical parkinsonian disorder, movement disorder of unknown aetiology or behavioural variant frontotemporal dementia (bv-FTD), four cases (3 %) showed positive staining for prion protein. Among neuropathologically classified non-tau FTLD cases (nine on fast-track, ten in total), one had the clinical diagnosis of CBD, one of LBD, one of PiD and of unclassified dementia with oral facial dystonia. The remaining cases were clinically classified as frontotemporal dementia (four cases), suspected bv-FTD or PSP (one case) and primary progressive aphasia (one case). The case with clinically suspected LBD showed a peculiar immunostaining with TDP-43 negative Sequestome-1 (p62) positive inclusions typical for C9ORF72 hexanucleotide expansion-related FTLD cases [4]), which prompted us to further assessment and final classification as FTLD-TDP. Additional cerebral amyloid angiopathy (CAA) occurred in 50 cases (37.6 %). We detected CAA on screening histology already in 46 cases (92 %). We found LBD co-pathology in 15 out of 37 AD cases (40.5 %) among which 13 cases where LBD co-pathology had already been seen on fast-track histology. Another frequent finding (12 / 37 cases, 32.4 % in our study) in AD is TDP-43 pathology, which was only identified after complete histopathological work-up in seven cases. Final histology revealed the presence of AD-associated alterations (Aβ plaques and / or AD-type tau deposits) in 15 / 23 LBD cases (65.2 %), 3/14 CBD cases (21.4 %), 4 / 10 non-tau FTLD cases (40 %), 9 / 14 MSA cases (64.3 %), 7 / 27 PSP cases (25.9 %). We detected signs of aging-related tau astrogliopathy (ARTAG) in seven LBD, three AD, three PSP, two FTLD non-tau and one CBD case. We reported ARTAG in five cases (3.8 %) on screening histology. Two AD, two CBD, eight LBD, one non-tau FTLD and one MSA case showed signs of argyrophilic grain disease (AGD) on final histology. Additional LBD pathology was present in one PSP and 15 AD cases upon examination of one hemisphere. Except for two cases (one with LBD Braak stage 5 and one with sole olfactory bulb pathology), all cases showed a Braak LBD stage 6 and could therefore be identified already on screening histology. Discussion In our study we demonstrated that a reliable, preliminary main diagnosis in patients with a suspected neurodegenerative disease can be achieved in over 96 % of the cases within a few days using histological and immunohistochemical screening of two selected, easily accessible brain regions (superior frontal gyrus and cerebellum) embedded into one paraffin block. Considering the rapid workflow, a prompt response from the neuropathologist to the clinician might improve the accuracy of clinical diagnosis in other patients. Furthermore, performing a screening approach might also be feasible in places with limited resources and could also be performed by general or forensic pathologists, perhaps with the aid of online consultation using digitalized micrographs. The fast-track diagnosis may also have consequences for relatives and lead to genetic counselling. However, our approach cannot supersede a thorough macro- and microscopic examination with sampling of various other brain regions, given the multitude of possible proteinopathic and vascular co-pathologies, potential extraordinarily rare NDD and alterations relevant for disease staging. By way of example, our approach neither allows for classification into brainstem-predominant, limbic or diffuse neocortical LB pathology according to the consensus report nor for determination of olfactory bulb only or amygdala predominant pathology [40,45,46]. Due to the heterogeneous spatial distribution pattern of subpial, subependymal, perivascular, white matter and gray matter ARTAG [47], it is possible that ARTAG pathology will be missed or not fully assessed by screening histology. Given the anatomical distribution of argyrophilic grain disease (AGD), often limited to the limbic system [48], assessment of this disease occurring frequently with concomitant NDD is limited with our method. This also holds true for the diagnosis of primary age-related tauopathy (PART) and LATE-NC which usually affects the middle frontal gyrus in later stages [49,50]. Furthermore, staging TDP-43 co-pathology in AD according to Josephs et al. [51] is not possible in our rapid screening approach. Independent from its diagnostic benefit, our procedure enhances laboratory safety by detecting unexpected prion protein aggregates before a complete work-up of the brain, as seen in four cases analysed. Since up-to-date cerebrospinal fluid based real-time quaking-induced conversion (RT-QuIC) has high sensitivity (> 90 %) and specificity (up to 100 %) for the detection of prion disease [52,53,54,55], an intravitam diagnosis in patients with suspected prion disease is possible. Yet, RT-QuIC might give false negative results as documented for instance in sporadic CJD cases at younger age or cases with VV1 or MM2 molecular subtype [52,56]. Patients with negative RT-QuIC are also more likely to present with motor symptoms or gait difficulties at initial presentation [56]. Additionally, implementing RT-QuIC assays is difficult and therefore often only a limited number of laboratories offer it [57]. Of note, absence of prion protein immunoreactivity in the fontal cortex and cerebellum does not entirely rule out the possibility of a prion disease since thalamic forms of Creutzfeldt-Jakob disease or fatal familial insomnia might not exhibit pathological prion protein aggregates in the frontal cortex or cerebellum [23,58]. Finally, in a different brain bank cohort, the use of two brain regions (frontal cortex and cerebellum) has been considered sufficient for the surveillance of prion disease [59]. The diagnostic accuracy of fast-track histology might even be higher by including temporomesial structures (hippocampus, amygdala) or thalamus (e.g. for the detection of rare prion disease variants). Diagnosing PART, as defined by tau NFT pathology with a Braak and Braak stage < IV and Thal plaque phases 0 - 2 [49], would require sampling of hippocampal structures including entorhinal and transentorhinal cortices as well as adjacent temporal neocortex. In particular, fast-track histology might enhance the recognition of common co-pathologies, such as AGD, ARTAG or LATE-NC which typically or often initiate in the limbic structures of the medial temporal lobe [47,48,50]. In our study however, handling and cutting of the brain was limited to a minimum for the sake of safety in respect to prion transmission and hence focused only on two easily accessible brain regions. Furthermore, our study was performed in a brain bank setting and all cases hence underwent a thorough neuropathological evaluation after screening histology, covering the aforementioned disease entities as well. Compared to the study by Venneti et al. [17] which assessed mid-frontal cortex (and additional cortical regions as well as basal ganglia) we achieved a higher diagnostic sensitivity. Remarkably that study [17] showed 0 % sensitivity for the diagnosis of PSP by applying only a frontal lobe specimen, whereas we reached a sensitivity of 96 %. In the study by King et al. [18], the sensitivity for the diagnosis of AD, CBD and MSA was 100 % for frontal specimens and frontal/temporal specimens combined, which is as high as in our study. However, we reached a higher sensitivity for the diagnosis of PSP (96 % vs. 0 %), LBD (100 % vs. 60 %) and FTLD non-tau (100 % vs. 83 % (combined) respectively 67 % (fontal sample alone). The authors of both previous studies [17,18] mention the sparse occurrence of frontal tau pathology in PSP and thus diagnostic difficulties. On the contrary, we found frontal tufted astrocytes in all cases and frontal coiled bodies in all but one case classified as PSP on screening histology. Since neuronal, astrocytic and oligodendroglial tau accumulation in the frontal cortex precedes pathology in other neocortical areas [60], we consider assessment of the superior frontal gyrus as useful for an orientating diagnosis of PSP. In a proposed stepwise distribution model of PSP, frontal tau can already be detected at stage 3, temporally just after anatomically less easily accessible regions of the subthalamic nucleus and basal ganglia [60]. Of note, we assessed brain tissue of participants in the frame of a brain bank program, essentially recruiting patients with a known history of a NDD and therefore often presenting with advanced disease and widespread pathology. This might for instance account for the high number of LBD cases showing frontal pathology. Conclusion We here developed a diagnostic algorithm based on the histomorphological assessment of two easily accessible brain regions to obtain a working diagnosis for the commonest NDD. Our diagnostic algorithm is highly reliable with an over 96 % concordance between the screening and final neuropathological diagnoses. In addition, it enhances the safety of laboratory personnel by excluding prion diseases before exhaustive manipulation of the brain. Furthermore, our diagnostic algorithm allows for a prompt feedback to the treating physicians. In conjunction with the clinical history, our screening diagnosis might foster a targeted evaluation of the fixed brain tissue. Finally, the approach presented here might be feasible in future biopsy diagnostics of NDD and thereby enhance the diagnostic rapidity of postmortem NDD diagnostics. Some caution is however warranted since our approach does not appreciate the manifold co-pathologies that may be present in NDD and since it has limited value for disease staging. Therefore, subsequent thorough analysis of various brain specimens is still required, especially in a brain bank setting. Acknowledgements We thank all brain donors and their relatives as well as the (neuro)pathologists involved in tissue asservation all over Germany for enabling this study. We further thank Mr. Michael Schmidt for excellent technical assistance. Conflicts of Interest Statement All authors do not disclose any conflict of interest. Funding Statement No funding was received to help prepare this manuscript. References 1. Tamsen, F. and I. Alafuzoff, When is a postmortem examination carried out? A retrospective analysis of all Swedish deaths 1999-2018. Virchows Arch, 2023. 482(4): p. 721-727. DOI: https://doi.org/10.1007/s00428-022-03462-w 2. Selvackadunco, S., et al., Comparison of clinical and neuropathological diagnoses of neurodegenerative diseases in two centres from the Brains for Dementia Research (BDR) cohort. J Neural Transm (Vienna), 2019. 126(3): p. 327-337. DOI: https://doi.org/10.1007/s00702-018-01967-w 3. Brunnstrom, H. and E. Englund, Clinicopathological concordance in dementia diagnostics. Am J Geriatr Psychiatry, 2009. 17(8): p. 664-70. DOI: https://doi.org/10.1097/jgp.0b013e3181a6516e 4. Shim, Y.S., et al., Clinicopathologic study of Alzheimer's disease: Alzheimer mimics. J Alzheimers Dis, 2013. 35(4): p. 799-811. DOI: https://doi.org/10.3233/JAD-121594 5. Osaki, Y., et al., Accuracy of clinical diagnosis of progressive supranuclear palsy. Mov Disord, 2004. 19(2): p. 181-9. DOI: https://doi.org/10.1002/mds.10680 6. el Tawil, S., et al., Variant Creutzfeldt-Jakob disease in older patients. J Neurol Neurosurg Psychiatry, 2015. 86(11): p. 1279-80. DOI: https://doi.org/10.1136/jnnp-2014-309397 7. Xiong, X., et al., Alzheimer's disease diagnostic accuracy by fluid and neuroimaging ATN framework. CNS Neurosci Ther, 2023. DOI: https://doi.org/10.1111/cns.14357 8. Kovacs, G.G. and H. Budka, Current concepts of neuropathological diagnostics in practice: neurodegenerative diseases. Clin Neuropathol, 2010. 29(5): p. 271-88. DOI: https://doi.org/10.5414/npp29271 9. Montine, T.J., et al., National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol, 2012. 123(1): p. 1-11. DOI: https://doi.org/10.1007/s00401-011-0910-3 10. Flanagan, M.E., et al., Performance of a Condensed Protocol That Reduces Effort and Cost of NIA-AA Guidelines for Neuropathologic Assessment of Alzheimer Disease. J Neuropathol Exp Neurol, 2017. 76(1): p. 39-43. DOI: https://doi.org/10.1093/jnen/nlw104 11. Clement, N.F., et al., A Simplified Brain Blocking Protocol Optimized for the Diagnosis of Neurodegenerative Disease Saves Time and Money While Preserving Anatomic Relationships. Arch Pathol Lab Med, 2021. 145(8): p. 960-968. DOI: https://doi.org/10.5858/arpa.2020-0322-OA 12. Maioli, H., et al., Performance of a condensed protocol to assess limbic-predominant age-related TDP-43 encephalopathy neuropathologic change. J Neuropathol Exp Neurol, 2023. 82(7): p. 611-619. DOI: https://doi.org/10.1093/jnen/nlad035 13. Multz, R.A., et al., What every neuropathologist needs to know: condensed protocol work-up for clinical dementia syndromes. J Neuropathol Exp Neurol, 2023. 82(2): p. 103-109. DOI: https://doi.org/10.1093/jnen/nlac114 14. Bharadwaj, R., et al., Application of the condensed protocol for the NIA-AA guidelines for the neuropathological assessment of Alzheimer's disease in an academic clinical practice. Histopathology, 2018. 72(3): p. 433-440. DOI: https://doi.org/10.1111/his.13345 15. Priemer, D.S. and R.D. Folkerth, Dementia in the Forensic Setting: Diagnoses Obtained Using a Condensed Protocol at the Office of Chief Medical Examiner, New York City. J Neuropathol Exp Neurol, 2021. 80(8): p. 724-730. DOI: https://doi.org/10.1093/jnen/nlab059 16. Lopez, A., et al., Flanagan's condensed protocol for neurodegenerative diseases. Implementation in a clinical autopsy setting with partial supervision of a neuropathologist. Virchows Arch, 2024. DOI: https://doi.org/10.1007/s00428-024-03781-0 17. Venneti, S., et al., Simulated brain biopsy for diagnosing neurodegeneration using autopsy-confirmed cases. Acta Neuropathol, 2011. 122(6): p. 737-45. DOI: https://doi.org/10.1007/s00401-011-0880-5 18. King, A., et al., Simulated surgical-type cerebral biopsies from post-mortem brains allows accurate neuropathological diagnoses in the majority of neurodegenerative disease groups. Acta Neuropathol Commun, 2013. 1: p. 53. DOI: https://doi.org/10.1186/2051-5960-1-53 19. Kitamoto, T., et al., Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest, 1987. 57(2): p. 230-6. 20. Hardt, M., T. Baron, and M.H. Groschup, A comparative study of immunohistochemical methods for detecting abnormal prion protein with monoclonal and polyclonal antibodies. J Comp Pathol, 2000. 122(1): p. 43-53. DOI: https://doi.org/10.1053/jcpa.1999.0343 21. Dickson, D.W., et al., Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol, 2002. 61(11): p. 935-46. DOI: https://doi.org/10.1093/jnen/61.11.935 22. Taipa, R., J. Pinho, and M. Melo-Pires, Clinico-pathological correlations of the most common neurodegenerative dementias. Front Neurol, 2012. 3: p. 68. DOI: https://doi.org/10.3389/fneur.2012.00068 23. Parchi, P., et al., Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol, 1999. 46(2): p. 224-33. 24. Hyman, B.T., et al., National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement, 2012. 8(1): p. 1-13. DOI: https://doi.org/10.1016/j.jalz.2011.10.007 25. Hoglinger, G.U., et al., Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord, 2017. 32(6): p. 853-864. DOI: https://doi.org/10.1002/mds.26987 26. Jecmenica Lukic, M., et al., Long-duration progressive supranuclear palsy: clinical course and pathological underpinnings. Ann Neurol, 2022. DOI: https://doi.org/10.1002/ana.26455 27. Dickson, D.W., Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol, 1999. 246 Suppl 2: p. II6-15. DOI: https://doi.org/10.1007/BF03161076 28. Saranza, G.M., et al., Corticobasal degeneration. Int Rev Neurobiol, 2019. 149: p. 87-136. DOI: https://doi.org/10.1016/bs.irn.2019.10.014 29. Uchihara, T. and K. Tsuchiya, Neuropathology of Pick body disease. Handb Clin Neurol, 2008. 89: p. 415-30. DOI: https://doi.org/10.1016/S0072-9752(07)01238-9 30. Alafuzoff, I., et al., Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol, 2009. 117(6): p. 635-52. DOI: https://doi.org/10.1007/s00401-009-0523-2 31. Walker, L., L. Stefanis, and J. Attems, Clinical and neuropathological differences between Parkinson's disease, Parkinson's disease dementia and dementia with Lewy bodies - current issues and future directions. J Neurochem, 2019. 150(5): p. 467-474. DOI: https://doi.org/10.1111/jnc.14698 32. Jellinger, K.A. and A.D. Korczyn, Are dementia with Lewy bodies and Parkinson's disease dementia the same disease? BMC Med, 2018. 16(1): p. 34. DOI: https://doi.org/10.1186/s12916-018-1016-8 33. Trojanowski, J.Q., T. Revesz, and M.S.A. Neuropathology Working Group on, Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol, 2007. 33(6): p. 615-20. DOI: https://doi.org/10.1111/j.1365-2990.2007.00907.x 34. Cairns, N.J., et al., Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol, 2007. 114(1): p. 5-22. DOI: https://doi.org/10.1007/s00401-007-0237-2 35. Pikkarainen, M., P. Hartikainen, and I. Alafuzoff, Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol, 2008. 67(4): p. 280-98. DOI: https://doi.org/10.1097/NEN.0b013e31816a1da2 36. Thal, D.R., et al., Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology, 2002. 58(12): p. 1791-800. DOI: https://doi.org/10.1212/wnl.58.12.1791 37. Braak, H., et al., Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol, 2006. 112(4): p. 389-404. DOI: https://doi.org/10.1007/s00401-006-0127-z 38. Thal, D.R., et al., Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol, 2002. 61(3): p. 282-93. DOI: https://doi.org/10.1093/jnen/61.3.282 39. Braak, H., et al., Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging, 2003. 24(2): p. 197-211. DOI: https://doi.org/10.1016/s0197-4580(02)00065-9 40. McKeith, I.G., et al., Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology, 2017. 89(1): p. 88-100. DOI: https://doi.org/10.1212/wnl.0000000000004058 41. Armstrong, M.J., et al., Criteria for the diagnosis of corticobasal degeneration. Neurology, 2013. 80(5): p. 496-503. DOI: https://doi.org/10.1212/WNL.0b013e31827f0fd1 42. Mackenzie, I.R., et al., Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol, 2009. 117(1): p. 15-8. DOI: https://doi.org/10.1007/s00401-008-0460-5 43. Landis, J.R. and G.G. Koch, The measurement of observer agreement for categorical data. Biometrics, 1977. 33(1): p. 159-74 44. Mann, D.M., et al., Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun, 2013. 1: p. 68. DOI: https://doi.org/10.1186/2051-5960-1-68 45. McKeith, I.G., et al., Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology, 2005. 65(12): p. 1863-72. DOI: https://doi.org/10.1212/01.wnl.0000187889.17253.b1 46. Beach, T.G., et al., Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol, 2009. 117(6): p. 613-34. DOI: https://doi.org/10.1007/s00401-009-0538-8 47. Kovacs, G.G., et al., Sequential stages and distribution patterns of aging-related tau astrogliopathy (ARTAG) in the human brain. Acta Neuropathol Commun, 2018. 6(1): p. 50. DOI: https://doi.org/10.1186/s40478-018-0552-y 48. Ferrer, I., G. Santpere, and F.W. van Leeuwen, Argyrophilic grain disease. Brain, 2008. 131(6): p. 1416-1432. DOI: https://doi.org/10.1093/brain/awm305 49. Crary, J.F., et al., Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol, 2014. 128(6): p. 755-66. DOI: https://doi.org/10.1007/s00401-014-1349-0 50. Nelson, P.T., et al., LATE-NC staging in routine neuropathologic diagnosis: an update. Acta Neuropathol, 2023. 145(2): p. 159-173. DOI: https://doi.org/10.1007/s00401-022-02524-2 51. Josephs, K.A., et al., Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol, 2016. 131(4): p. 571-85. DOI: https://doi.org/10.1007/s00401-016-1537-1 52. Watson, N., et al., Validation of Revised International Creutzfeldt-Jakob Disease Surveillance Network Diagnostic Criteria for Sporadic Creutzfeldt-Jakob Disease. JAMA Netw Open, 2022. 5(1): p. e2146319. DOI: https://doi.org/10.1001/jamanetworkopen.2021.46319 53. Mastrangelo, A., et al., Evaluation of the impact of CSF prion RT-QuIC and amended criteria on the clinical diagnosis of Creutzfeldt-Jakob disease: a 10-year study in Italy. J Neurol Neurosurg Psychiatry, 2023. 94(2): p. 121-129. DOI: https://doi.org/10.1136/jnnp-2022-330153 54. Bsoul, R., et al., Improved Real-Time Quaking Induced Conversion for Early Diagnostics of Creutzfeldt-Jakob Disease in Denmark. Int J Mol Sci, 2023. 24(7). DOI: https://doi.org/10.3390/ijms24076098 55. Hermann, P., et al., Application of real-time quaking-induced conversion in Creutzfeldt-Jakob disease surveillance. J Neurol, 2023. 270(4): p. 2149-2161. DOI: https://doi.org/10.1007/s00415-022-11549-2 56. Ng, D., et al., Characterisation of RT-QuIC negative cases from the UK National CJD Research and Surveillance programme. J Neurol, 2024. DOI: https://doi.org/10.1007/s00415-024-12345-w 57. Hermann, P., et al., Validation and utilization of amended diagnostic criteria in Creutzfeldt-Jakob disease surveillance. Neurology, 2018. 91(4): p. e331-e338. DOI: https://doi.org/10.1212/WNL.0000000000005860 58. Zerr, I., et al., Phenotypic variability in fatal familial insomnia (D178N-129M) genotype. Neurology, 1998. 51(5): p. 1398-405. DOI: https://doi.org/10.1212/wnl.51.5.1398 59. Peden, A.H., et al., Enhanced Creutzfeldt-Jakob disease surveillance in the older population: Assessment of a protocol for screening brain tissue donations for prion disease. Brain Pathol, 2023: p. e13214. DOI: https://doi.org/10.1111/bpa.13214 60. Kovacs, G.G., et al., Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol, 2020. 140(2): p. 99-119. DOI: https://doi.org/10.1007/s00401-020-02158-2
Copyright: © 2024 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |