|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Free Neuropathology 5:11 (2024) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Case Report |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Malignant glioma in L-2-Hydroxyglutaric Aciduria: thorough molecular characterization of a case and literature review |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Fleur Cordier1,2, Pieter Wesseling1,3, Bastiaan B.J. Tops1, Lennart Kester1, Pim J. French4, Martin van den Bent4, Felix Hinz5,11, Eleonora Aronica3, K. Mariam Slot6, Floor Abbink7, Marjo S. van der Knaap8,9,10, Mariëtte E.G. Kranendonk1 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Corresponding authors: |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Submitted: 15 February 2024 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Keywords: L-2-hydroxyglutaric aciduria, CNS tumor, Paediatric-type diffuse high-grade glioma, DNA-methylation-classification, Sequencing |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
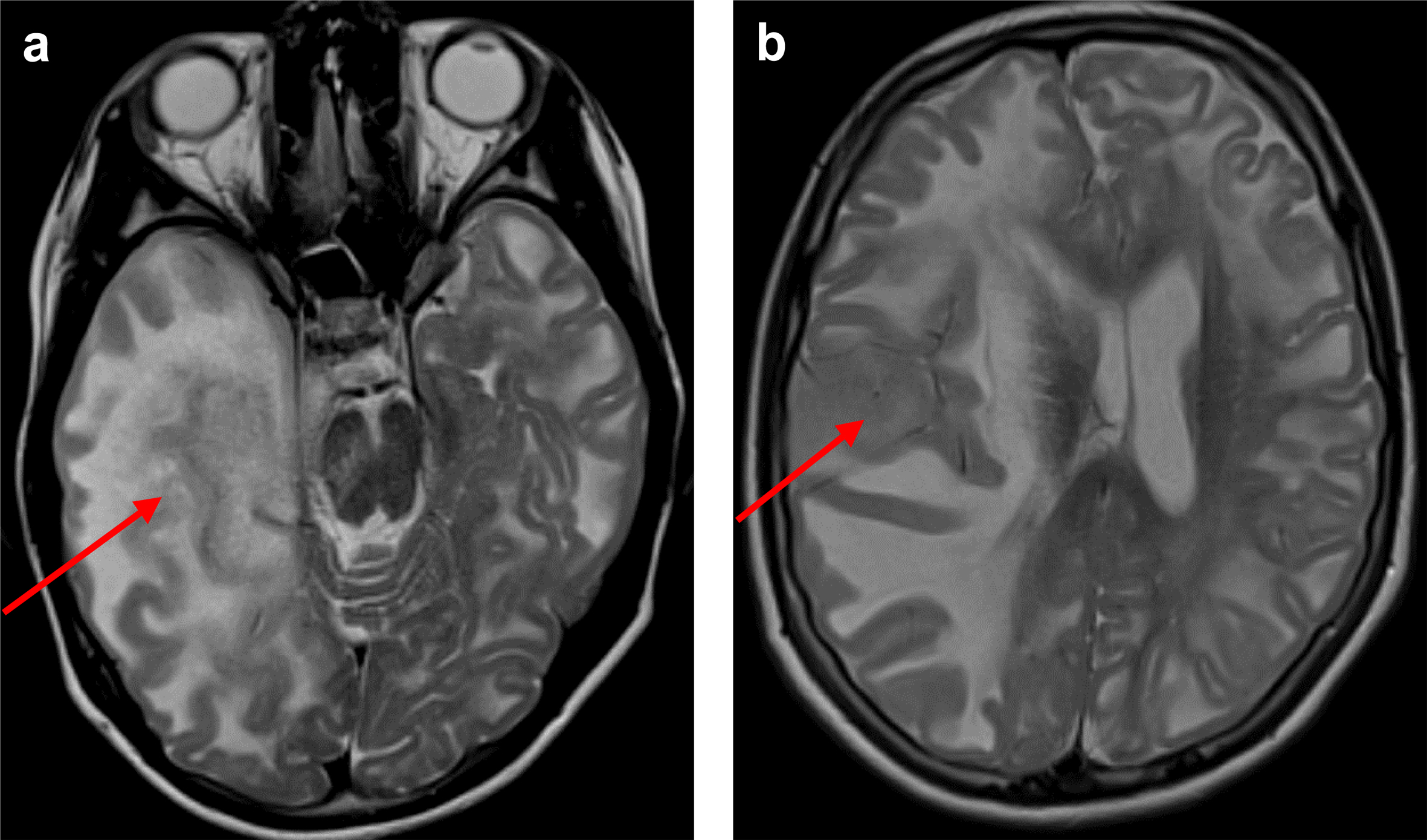
AbstractL-2-hydroxyglutaric aciduria (L-2-HGA) is a rare neurometabolic disorder characterized by accumulation of L2-hydroxyglutarate (L-2-HG) due to mutations in the L2HGDH gene. L-2-HGA patients have a significantly increased lifetime risk of central nervous system (CNS) tumors. Here, we present a 16-year-old girl with L-2-HGA who developed a tumor in the right cerebral hemisphere, which was discovered after left-sided neurological deficits of the patient. Histologically, the tumor had a high-grade diffuse glioma phenotype. DNA sequencing revealed the inactivating homozygous germline L2HGDH mutation as well as inactivating mutations in TP53, BCOR and NF1. Genome-wide DNA-methylation analysis was unable to classify the tumor with high confidence. More detailed analysis revealed that this tumor clustered amongst IDH-wildtype gliomas by methylation profiling and did not show the glioma CpG island methylator phenotype (G-CIMP) in contrast to IDH-mutant diffuse gliomas with accumulated levels of D-2-HG, the stereoisomer of L-2-HD. These findings were against all our expectations given the inhibitory potential of 2-HG on DNA-demethylation enzymes. Our final integrated histomolecular diagnosis of the tumor was diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype. Due to rapid tumor progression the patient died nine months after initial diagnosis. In this manuscript, we provide extensive molecular characterization of the tumor as well as a literature review focusing on oncogenetic considerations of L-2-HGA-associated CNS tumors. IntroductionL-2-hydroxyglutaric aciduria (L-2-HGA) is an autosomal recessive neurometabolic disorder.1 The disease is caused by pathogenic variants in the L2HGDH gene (14q22.1) encoding L-2-hydroxyglutarate dehydrogenase, a flavin adenine dinucleotide (FAD)-dependent enzyme that normally oxidizes L-2-hydroxyglutarate (L-2-HG) to alpha-ketoglutarate. Defective enzyme activity leads to L-2-HG accumulation with a myelinotoxic and oncogenic effect on the central nervous system (CNS) and L-2-HG accumulation in urine, plasma, and cerebrospinal fluid.2 The disease poses a relatively consistent pattern of presentation, starting with delayed mental and motor development in the first years of life, epilepsy in around 60 % of the cases, continuing with a slowly progressive course eventually leading to ataxia and moderate to severe mental deterioration.2,3,4,5,6,7 By imaging, a unique pattern of abnormalities is seen, characterized by subcortical leukoencephalopathy with relative sparing of the periventricular white matter, corpus callosum, cerebellar white matter and the brainstem tracts. Additional signal alterations in the basal ganglia and dentate nuclei are typical.2,8,9 A minority of patients also develop a CNS tumor, usually later in life.2 Because of the enhanced tumor risk, regular imaging for patients with L-2-HGA is part of standard care, typically once per 1 or 2 years. Here we present a teenager with L-2-HGA who developed a glial neoplasm in the cerebral hemisphere. We hypothesized that the oncogenesis of this tumor would be in line with IDH-mutant astrocytomas where D-2-HG (the enantiomer of L-2-HG) is considered the oncometabolite leading to the glioma CpG island methylated phenotype (G-CIMP).10,11 However, the tumor of the L-2-HGA patient did not show G-CIMP and was challenging-to-classify, even using a combination of state of the art molecular diagnostic tools. Additionally, we provide an overview of L-2-HGA-associated CNS tumors reported in the literature and discuss the similarities and differences with IDH-mutant gliomas. CaseA 16-year-old girl, the sole child of a consanguineous couple, experienced a normal early development and could walk without support at 13 months. Speech development was delayed, and she exhibited mild motor coordination issues. At 5 years age, an MRI revealed extensive subcortical cerebral white matter abnormalities and signal abnormalities in the basal ganglia and dentate nuclei, characteristic of L-2-HGA. The diagnosis was confirmed through elevated urinary excretion of L-2-HG and the identification of a homozygous variant in the L2HGDH gene (L2HGDH(NM_024884.2): c.339T > A p.(Cys113*)).12 The parents were confirmed to be heterozygous for the same variant. Over the years, the patient developed moderate cognitive impairment and mild cerebellar ataxia. Starting at the age of 14, she experienced occasional epileptic seizures, treated with Valproate. MRI at the age of 14 years did not show evidence of tumor development. At age 16, a new MRI, prompted by a new-onset subtle left-sided central paresis, revealed an augmentation in T2 hyperintensity and swelling in most of the right cerebral hemisphere. The border between cortex and subcortical white matter was blurred, particularly medial in the temporal lobe, where the cortex was thickened and had a slightly increased T2 signal (Figure 1a). A similar tumorous area was identified in the right superior temporal gyrus (Figure 1b). Distinguishing pre-existing abnormal white matter related to L-2-HGA from the new abnormalities within the white matter was challenging. The signal abnormalities extended into the right thalamus, right cerebral peduncle, pons, and were accompanied by subfalcine herniation and a midline shift to the left (Figure 1a,b). Post-contrast scans did not reveal abnormal enhancement. Several small foci of diffusion restriction were present within the tumor. Based on the findings in serial MRI examinations a radiological diagnosis of a rapidly growing, likely high-grade glial tumor was made. A biopsy of the tumor was taken from the right temporal lobe near the superior temporal gyrus to confirm its nature.
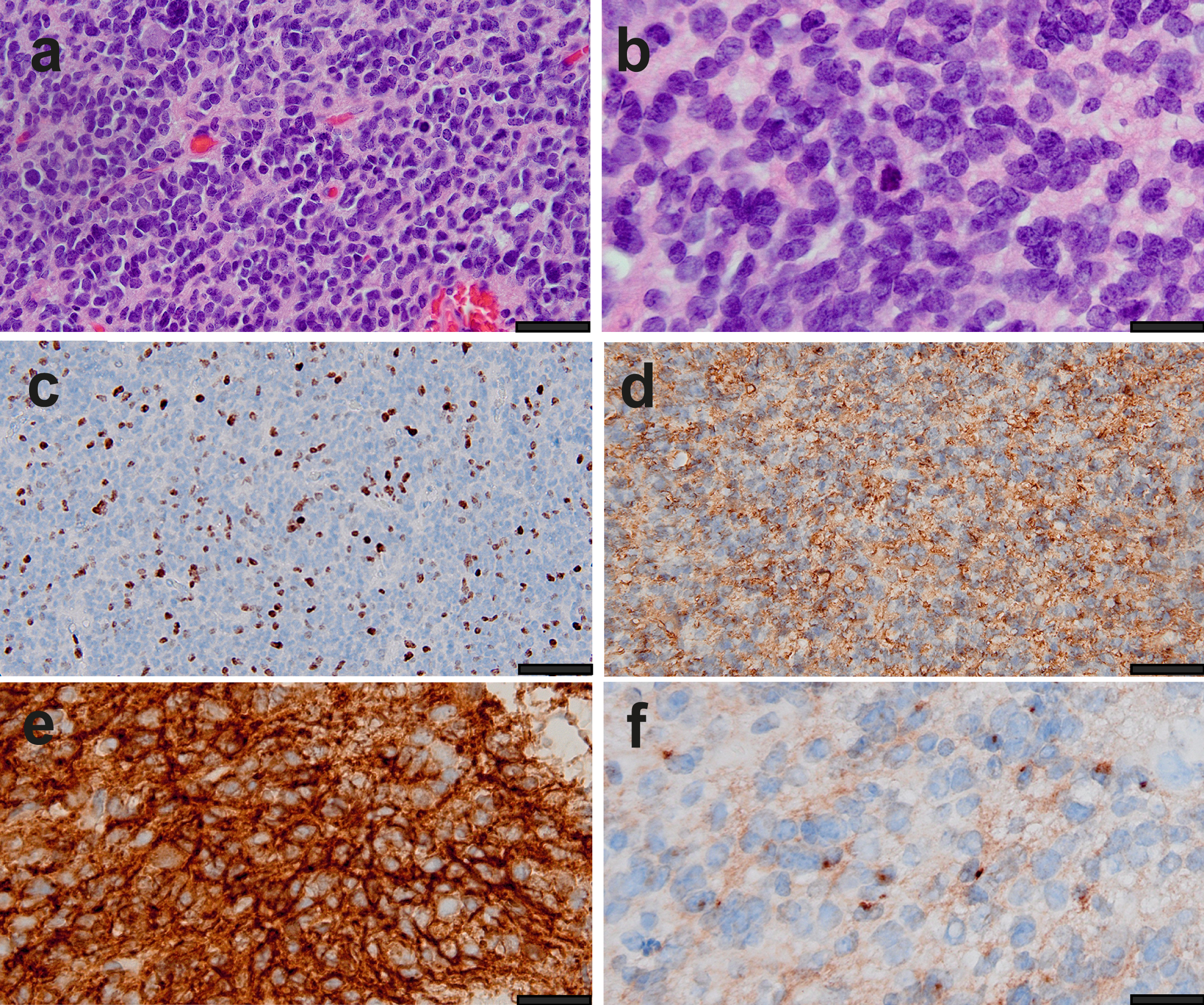
Figure 1. Preoperative MRI. Axial T2-weighted MR images with the red arrows indicating a space occupying lesion in the medial part of the right temporal lobe (a) and in the right superior temporal gyrus (b, i.e. the location of the biopsy), accompanied by midline shift and brain herniation (transtentorial in a, subfalcine in b). Additionally, in both hemispheres T2 hyperintensity of the white matter is present, pathognomonic for L2-HGA. HistologyHistological analysis revealed a tumor consisting of small, poorly differentiated appearing cells with diffuse infiltrative growth in the preexistent brain parenchyma. The tumor cell nuclei were round to ovoid, showed substantial atypia and fine-grained chromatin with small nucleoli. Mitotic figures were frequent. Notably, the cells showed some degree of clustering with small areas of pre-existent neuropil in between clusters, but lacked true rosettes or evident perivascular pseudorosettes (Figure 2a,b). Florid microvascular proliferation and necrosis were absent.
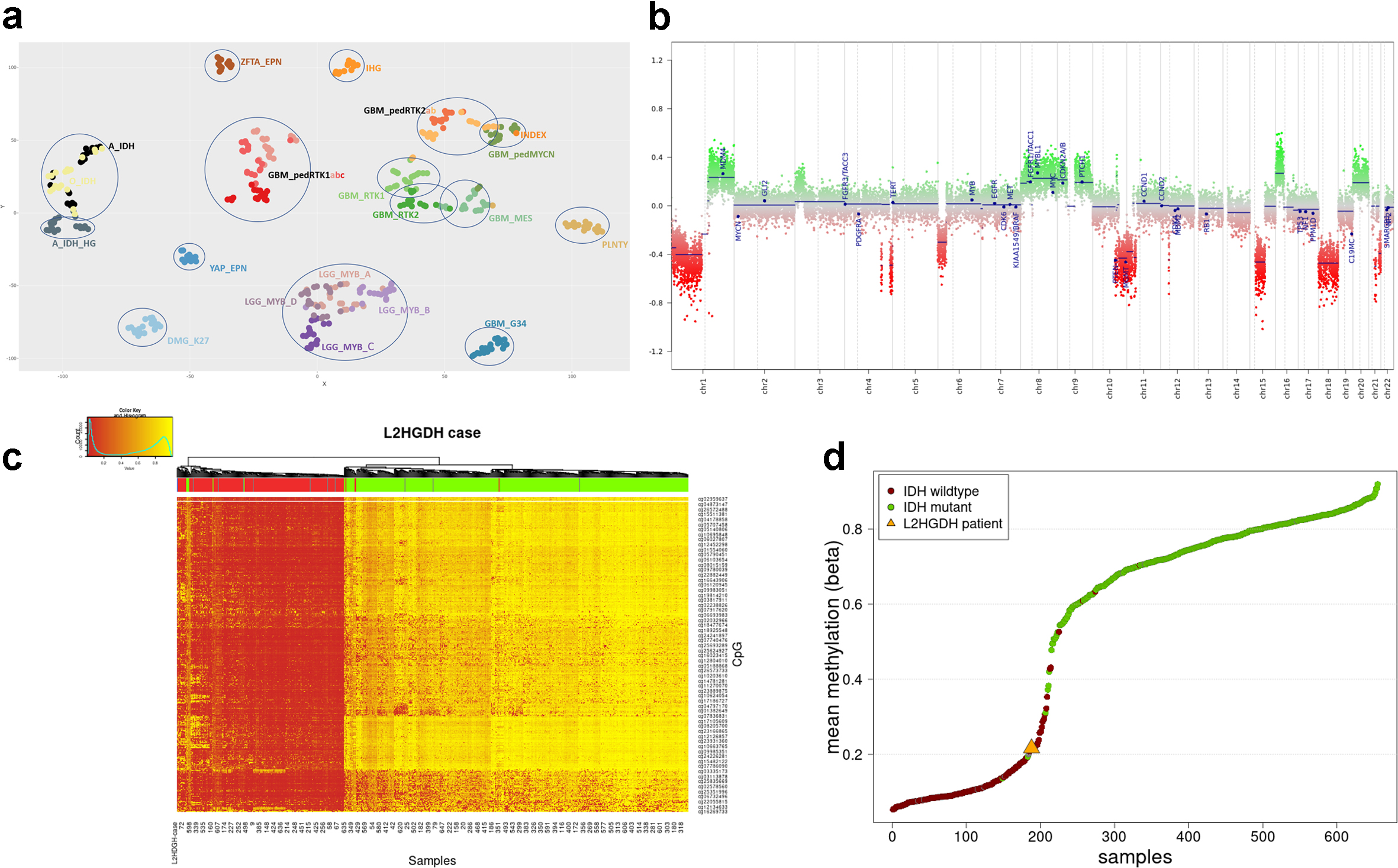
Figure 2. Histological and immunohistochemical characteristics of the tumor. (a,b) Low and high magnification of the diffuse infiltrative high-grade glioma. (c-f) Immunoreactivity for Ki-67 (c), GFAP (d), MAP2 (e) and EMA (f). Scale bars: 80 um (a,c,d); 50 um (b,e,f). Immunohistochemically, the tumor cells were positive for vimentin, CD56, and MAP2, and in some areas for synaptophysin and GFAP as well. Dot-like EMA staining was found in a small number of tumor cells. The tumor cells were negative in the following stainings: CAM5.2, CD34 (with positive control in the vessels), NeuN (staining only pre-existing neurons), Olig2, IDH1 R132H and H3 p.K28M. Furthermore, in the H3 p.K28me3, ATRX, and INI-1 staining the nuclear expression was retained. The Ki-67 labelling index was variable but locally >= 30 %. (Figure 2c-f). Molecular findingsDNA methylation analysis was performed using the Infinium MethylationEpic beadchip (Illumina), and explored using versions 11b4, 12.5 and 12.8 of the Heidelberg classifier developed by DKFZ (https://www.molecularneuropathology.org/mnp/)8 as well as the Bethesda classifier.13 Neither of these classifiers were able to classify the tumor with a high confidentiality score. The best classification possible was obtained with DKFZ v12.8 which classified the tumor as pediatric-type diffuse high-grade glioma with a highest score of 0.83, diffuse Paediatric Type High Grade Glioma, Mycn Subtype (calibrated score 0.52). The Bethesda classifier and UMAP (Uniform Manifold Approximation and Projection) suggested IDH-mutant glioma with a score of 0.54. Based on DNA methylation-based t-distributed stochastic neighbor embedding (t-SNE) clustering analyses, this tumor clustered most close to pediatric-type diffuse high-grade glioma, MYCN-activated subtype (Figure 3a).
Figure 3. DNA methylation analysis of the tumor. (a) DNA methylation-based t-distributed stochastic neighbor embedding (tSNE) of the tumor of our patient with L-2-HGA (marked as INDEX, see arrow) compared to selected reference samples from the Heidelberg database (see supplemental data for abbreviations). The tSNE was run with following settings: CpGs analysed = 10.000, iterations = 3.000, perplexity = 5, theta = 0, dims = 2. Our case was in close proximity to MYCN-activated pediatric high-grade gliomas. Samples are colored according to their methylation class. (b) Copy number plot showing a rather complex profile with multiple chromosomal gains and losses. (c,d) Methylation status in comparison to IDH-mutant gliomas. Unsupervised analysis using the 1000 most variable probes of a cohort of 435 IDH-mutant and 215 IDH-wildtype gliomas15 revealed that the tumor of our patient clustered amongst the IDH-wildtype tumors (Figure 3c, color-coding of the samples: IDH-mutant, IDH-wildtype and unknown samples are indicated respectively in green, red and grey. The index case is indicated in blue and by the red rectangle). (Figure 3d, Mean methylation beta value of the 1000 most variable probes shown in c. The IDH-wildtype cases are colored in red, the IDH-mutant cases in green and the index sample is highlighted with an orange triangle.) The copy number variation (CNV) plot obtained by the conumee package embedded in the methylome profiling analysis and whole exome sequencing (WES) revealed a complex CNV profile with multiple gains and losses, including partial loss of chromosome 10q. There was no high-level amplification of EGFR or MYCN, no chromosome 7 gain, no homozygous deletion of CDKN2A/B and no chromosome 1p/19q codeletion (Figure 3b). The promoter of the O6-methylguanine-DNA methyltransferase (MGMT) gene was found to be methylated. WES identified somatic mutations in TP53 (c.818G > A (p.Arg273His)); 96.6 % VAF (including copy neutral LOH), NF1:c.4977_4980delCTCT (p.Lys1661Glyfs*36); 95.1 % VAF (including copy neutral LOH) and BCOR:c.4438C > T (p.Arg1480*); 93.7 % VAF (including copy neutral LOH). Furthermore, the known homozygous germline mutation in L2HGDH (c.339T > A p.(Cys113*)) was detected in the tumor as well (100 % VAF). RNA sequencing revealed no additional mutations or gene rearrangements. RNA levels of L2HGDH in the tumor were comparable to or even lower than in other pediatric tumors in the reference cohort of the princess Maxima center (Supplemental Figure 1). Unfortunately, there was no tissue left for analysis of L2HGDH protein levels in the tumor. Acknowledging that in IDH-mutant tumors high levels of D-2-HG (the enantiomer of L-2-HG) result in hypermethylation and G-CIMP9, 14, we investigated whether CIMP was present in the tumor of our patient with L-2-HGA as well. However, unsupervised analysis using the 1000 most variable probes of a cohort of 435 IDH-mutant and 215 IDH-wildtype gliomas15 revealed that the tumor of our patient clustered amongst the IDH-wildtype tumors (Figure 3c) and had relative low methylation levels (Figure 3d). Follow upGiven the dismal prognosis, which was partly attributed to the large size of the tumor, a restrained treatment policy was chosen. Following the ACNS 0126 protocol, the patient began treatment with Temodal two months after diagnosis, for stabilization of the tumor, in addition to dexamethasone for edema relief. Unfortunately, in the course of the following months the condition of the patient rapidly worsened, leading to her death nine months after the diagnosis of the CNS tumor. Literature review of L-2-HGA related CNS tumorsThe association of L-2-HGA and CNS tumors has been documented in the literature, with an estimated rate of association of 5 % to 40 %2 according to available data (see Table 1 for a compilation of cases in the literature). The spectrum of CNS tumors reported to be associated with L-2-HGA includes medulloblastoma in early childhood, anaplastic ependymoma predominantly in teenagers, low- and high-grade gliomas in both childhood and adults, and embryonal tumors (previously designated as “primitive neuroectodermal tumor”/PNETs). Apart from medulloblastomas (which by definition arise in the posterior fossa), L-2-HGA tumors are often located in a cerebral hemisphere (frontal lobe, temporal lobe and/or thalamus), but rarely in the hippocampus or in the ventricles. However, there is some missing data in the cases described in the literature: in one case the tumor diagnosis is not described, in three cases the age is lacking, in four cases information regarding the sex is not reported and most cases do not entail survival data. It is worth noting that translating previously assigned diagnoses for CNS tumors associated with L-2-HGA (Table 1) into labels conforming to WHO CNS 5 criteria poses some challenges.16,17 For example, the tumors of patients 13 and 20 in Table 1 were diagnosed as glioblastoma, while according to the WHO CNS 5 classification they would likely be classified as diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype. Additionally, no IDH status was reported in the astrocytoma or oligodendroglioma cases, which is an essential piece of information for a correct diagnosis of diffuse gliomas nowadays. Also, the case regarded as ependymoma and even the embryonal tumors may turn out to be high grade gliomas.18 All in all, it is therefore difficult to associate a specific tumor type with L-2-HGA. DiscussionIn this report, we present a teenager with L-2-HGA who developed a temporal lesion, histomolecularly classified as diffuse pediatric-type high-grade glioma, H3-wildtype, and IDH-wildtype according to the WHO CNS 5 criteria. This tumor entity represents a group of malignant diffuse gliomas, primarily occurring in children or young adults that lack alterations in histone H3 genes, as well as in IDH1 and IDH2. The molecular alterations in this group of tumors are diverse and include TP53 mutations, MYCN amplification, PDGFRA, ID2, NF1 or EGFR alterations. By DNA methylation profiling they comprise a distinct group with several molecular subgroups (RTK1, RTK2 and MYCN).16,19 Methylation array classification is based on “fingerprinting” the methylome of the tumor, and subsequently mapping this to a reference database.8 This methylome profile is a combination of both somatically acquired DNA methylation changes as well as the methylation status of the cell of origin. Noteworthy, high-grade gliomas based on a tumor predisposition syndrome such as mismatch repair (MMR) deficiency or Li-Fraumeni are often classified as diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype as well, although not always with high confidence. Capper et al. described that the full spectrum of tumors in the context of a hereditary predisposition syndrome may not yet be reliably classified.20 In line with this, the tumor we present here could also not be classified with confidence. A possible explanation for classification difficulties might be that the effect of the germline alteration, which is present in all cells, together with the somewhat different oncogenesis alters the methylome of the cancer in such a way that it no longer fully reflects the methylome profile of the sporadic counterparts in the reference cohort. It remains to be seen if tumors that arise in the context of L-2-HGA are (also on the epigenetic level) more similar to each other than suggested based on literature in the pre-molecular era (Table 1). Methylation analysis of tumors from additional L2-HGA patients is necessary for further characterization of L2-HGA-associated tumors, and for corroborating the absence of G-CIMP that we observed in our case. Table 1: Reported CNS tumors associated with L-2-HGA
M: male, F: female, NM: not mentioned, ND: not determined, PNET: primitive neuroectodermal tumor, FU: follow-up Interestingly, IDH-mutant diffuse gliomas exhibit elevated levels of D-2-hydroxyglutarate (D-2-HG), i.e. the enantiomer of the L-2-HG.21 This (onco) metabolite is a competitive inhibitor of alpha ketoglutarate-dependent dioxygenases, including the Ten-Eleven Translocation (TET) enzymes which play a crucial role in the initial stages of DNA demethylation. Consequently, high levels of D-2-HG in IDH-mutated tumors lead to hypermethylation and the manifestation of G-CIMP. Notably, D-2-HG alone is sufficient to induce such a CIMP phenotype.9,14 Similarly, L-2-HG inhibits TET1 and TET2 enzymes,2,11,22 and elevated levels of L-2HG were associated with hypermethylation of DNA in clear cell renal cell carcinoma.23 Even more, it has been shown that L-2-HG was a more potent inhibitor of both TET2 and TET1 than D-2-HG.24 Based on these findings, we hypothesize that individuals with L-2-HGA are at risk of developing diffuse glial tumors due to the pathological accumulation of L-2-HG in the brain, with a similar G-CIMP impact on glial cells and maybe even clustering with the IDH-mutant glioma cluster.2,21,25 However, in comparison with the methylation array data of 650 adult-type diffuse gliomas of the CATNON database, the tumor of our patient showed relatively low mean DNA methylation levels at CpG sites, and (in line with most methylation classifier and WES results) clustered with IDH-wildtype samples. This unexpected observation illustrates the complexity of elevated levels of D-2-HG versus L-2-HG in oncogenesis. One possible explanation is that the mechanism underlying elevated levels of 2HG differs between L2HGDH- or L2HGDH-associated disease compared to IDH-mutant tumors. Mutations in L2HGDH or D2HGDH lead to a generalized passive accumulation of 2HG throughout the body, whereas mutations in IDH genes in tumors result in high levels of 2HG within the tumor cells themselves with therefore possibly stronger local effect on TET-enzyme activity and subsequent hypermethylation.26,27 Of note, elevated D-2-HG levels can also occur in the context of neurometabolic disorders D-2-HGA type 1 (due to a mutation in the D2HGDH gene) and type 2 (due to heterozygous mutation in IDH2). In that context, increased D-2-HG levels do not lead to gliomas.28 This can be partly attributed to discrepancies in life expectancy, as patients with type 2 D-2-HGA, based on an IDH2 mutation, exhibit similarly elevated D-2-HG levels but have such a severe clinical course that they may not survive long enough for glioma development.29,30,31 Another explanation can be sought in stereoisomerism. Even though L-2-HG and D-2-HG are structurally similar molecules, studies have suggested that L-2-HG and D-2-HG may have differential inhibitory effects on certain enzymes, conceivably due to variations in the stereochemistry of L-2-HG and D-2-HG molecules. Overall, while both L-2-HG and D-2-HG function as inhibitors in cellular metabolism, their specific effects and potency may vary depending on the context and the particular enzymes or pathways involved. This is further stressed by the observation that the level of L-2-HG does not per se correlate with risk of CNS tumor development.32 Therefore it seems that 2-HG has physiological roles that extend beyond being an oncometabolite by itself. Notably, there appears to be no sex predisposition to the carcinogenic effect.2 Also, so far, evidence for a common molecular mechanism downstream of the L2HGDH mutation is lacking. Patay et al. (2015) identified an EGFR amplification in their case of anaplastic astrocytoma (Table 1, case 19) which lacked an IDH mutation. They therefore proposed a potential connection between astrocytic tumors and EGFR alterations as potential oncogenic driver mechanism in L-2-HG-induced tumorigenesis.21 However, in our case, and in all other cases presented in the literature, no EGFR alterations were reported. Possibly, the oncogenesis in L-2-HGA is like a two-tiered oncogenesis system in which accumulated levels of L-2-HG form the first hit, and a mutation (which may arise in different pathways) forms the second hit eventually leading to tumor formation, with a different pathophysiology than in the IDH-mutant tumor. Besides the L2HGDH mutation, the tumor analyzed in this report also contained TP53, BCOR and NF1 mutations, as well as numerous copy number changes. It is known that malignant progression of IDH-mutant astrocytomas is associated with relative hypomethylation (reduced G-CIMP status), a phenomenon which may be associated with (homozygous) CDKN2A/B loss or other cell cycle gene alterations in these tumors.33,34 However, in the tumor of our L-2-HGA patient homozygous loss of CDKN2A/B was absent. We speculate that in our case other molecular alterations had an impact on L-2-HG levels, TET-enzymes or other epigenetic mechanisms resulting in absence of G-CIMP. Unfortunately, no tumor tissue was left for proper assessment of L-2-HG at the protein level. Taken together, our findings shed unprecedented light on the unique developmental and epigenetic roots of a L-2-HGA associated glioma. Further studies are necessary to unravel how exactly the oncogenetic mechanism of elevated L-2-HG is different from that of its enantiomer D-2-HG. Based on the literature, the prognosis for L-2-HGA patients suffering from a CNS tumor appears to be poor. Among the documented cases, the patients experienced death with a mean survival of 10.8 months after diagnosis, although this is based on limited data. This poor outcome may partly be explained by delay in the diagnosis of these tumors, e.g., because on MRI scans the tumors are often not clearly delineated and/or difficult to distinguish from diseased non-neoplastic brain tissue. Additionally, among the reported patients, one individual (case 19) was described with a second tumor diagnosed 40 months after initial diagnosis and with the same histological and molecular characteristics as the first.21 ConclusionThis is the first report of a comprehensive molecular analysis of a challenging-to-classify CNS neoplasm in a teenager known to suffer from L-2-HGA. Intriguingly, we found that elevated levels of L-2-HG resulted in a tumor with characteristics of IDH-wildtype rather than of IDH-mutant diffuse gliomas, including a lack of G-CIMP in the tumor of our patient. Our study thereby illustrates that the oncogenic mechanism of increased L-2-HG levels in diffuse gliomas is different from that caused by D-2-HG accumulation in IDH-mutant diffuse gliomas. AcknowledgmentsWe kindly acknowledge Prof. Ken Aldape and Zied Abdullaev for providing methylation classification results of the tumor of our patient using the Bethesda (NIH/NCI) methylation classifier. Author contributionsFC, PW, PJF, MSvdK, and MEGK established the study concept and design. FC, PW and MEGK performed the writing of the paper. PJF, EA, FH, KMS, MSvdK and MEGK contributed the figures. All authors contributed essential ‘building blocks’ for this paper and have read, edited and approved the final version of the manuscript. Competing interestsThe authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. References1. Duran M, Kamerling JP, Bakker HD, van Gennip AH, Wadman SK. L-2-Hydroxyglutaric aciduria: an inborn error of metabolism? Journal of inherited metabolic disease 3, 109-112 (1980). https://doi.org/10.1007/BF02312543 2. Patay Z, Mills JC, Lobel U, Lambert A, Sablauer A, Ellison DW. Cerebral neoplasms in L-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data. AJNR American journal of neuroradiology 33, 940-943 (2012). https://doi.org/10.3174/ajnr.A2869 3. Barth PG, et al. L-2-hydroxyglutaric acidemia: a novel inherited neurometabolic disease. Annals of neurology 32, 66-71 (1992). https://doi.org/10.1002/ana.410320111 4. Goffette SM, Duprez TP, Nassogne MC, Vincent MF, Jakobs C, Sindic CJ. L-2-Hydroxyglutaric aciduria: clinical, genetic, and brain MRI characteristics in two adult sisters. European journal of neurology 13, 499-504 (2006). https://doi.org/10.1111/j.1468-1331.2006.01282.x 5. Moroni I, et al. L-2-hydroxyglutaric aciduria and brain malignant tumors: a predisposing condition? Neurology 62, 1882-1884 (2004). https://doi.org/10.1212/01.wnl.0000125335.21381.87 6. Rzem R, et al. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proceedings of the National Academy of Sciences of the United States of America 101, 16849-16854 (2004). https://doi.org/10.1073/pnas.0404840101 7. Steenweg ME, et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotype-phenotype study. Human mutation 31, 380-390 (2010). https://doi.org/10.1002/humu.21197 8. Capper D, et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469-474 (2018). https://doi.org/10.1038/nature26000 9. Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ. Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? Lancet Oncol 12, 83-91 (2011). https://doi.org/10.1016/S1470-2045(10)70053-X 10. Turcan S, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 4, 1729-1736 (2013). https://doi.org/10.18632/oncotarget.1412 11. Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell 17, 510-522 (2010). https://doi.org/10.1016/j.ccr.2010.03.017 12. den Dunnen JT, et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Human mutation 37, 564-569 (2016). https://doi.org/10.1002/humu.22981 13. Wu Z, et al. Impact of the methylation classifier and ancillary methods on CNS tumor diagnostics. Neuro-oncology 24, 571-581 (2022). https://doi.org/10.1093/neuonc/noab227 14. Cahill D, Turcan S. Origin of Gliomas. Semin Neurol 38, 5-10 (2018). https://doi.org/10.1055/s-0037-1620238 15. van den Bent MJ, et al. Interim results from the CATNON trial (EORTC study 26053-22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: a phase 3, randomised, open-label intergroup study. Lancet (London, England) 390, 1645-1653 (2017). https://doi.org/10.1016/S0140-6736(17)31442-3 16. WHO Classification of central nervous tumors Tumors, 5 edn. International Agency for Research on Cancer (2021). ISBN = 9789283245087 17. Mackay A, et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer cell 32, 520-537 e525 (2017). https://doi.org/10.1016/j.ccell.2017.08.017 18. Sturm D, et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164, 1060-1072 (2016). https://doi.org/10.1016/j.cell.2016.01.015 19. Thomas DL. 2021 updates to the World Health Organization classification of adult-type and pediatric-type diffuse gliomas: a clinical practice review. Chinese clinical oncology 12, 7 (2023). https://doi.org/10.21037/cco-22-120 20. Capper D, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta neuropathologica 136, 181-210 (2018). https://doi.org/10.1007/s00401-018-1879-y 21. Patay Z, et al. Successive distinct high-grade gliomas in L-2-hydroxyglutaric aciduria. Journal of inherited metabolic disease 38, 273-277 (2015). https://doi.org/10.1007/s10545-014-9782-8 22. Gunn K, et al. (R)-2-Hydroxyglutarate Inhibits KDM5 Histone Lysine Demethylases to Drive Transformation in IDH-Mutant Cancers. Cancer Discov 13, 1478-1497 (2023). https://doi.org/10.1158/2159-8290.CD-22-0825 23. Shelar S, et al. Biochemical and Epigenetic Insights into L-2-Hydroxyglutarate, a Potential Therapeutic Target in Renal Cancer. Clin Cancer Res 24, 6433-6446 (2018). https://doi.org/10.1158/1078-0432.CCR-18-1727 24. Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell 19, 17-30 (2011). https://doi.org/10.1016/j.ccr.2010.12.014 25. London F, Jeanjean A. Gliomatosis cerebri in L-2-hydroxyglutaric aciduria. Acta neurologica Belgica 115, 749-751 (2015). https://doi.org/10.1007/s13760-015-0489-x 26. Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739-744 (2009). https://doi.org/10.1038/nature08617 27. Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nature reviews Clinical oncology 18, 645-661 (2021). https://doi.org/10.1038/s41571-021-00521-0 28. Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis 35, 571-587 (2012). https://doi.org/10.1007/s10545-012-9462-5 29. Kranendijk M, et al. Evidence for genetic heterogeneity in D-2-hydroxyglutaric aciduria. Hum Mutat 31, 279-283 (2010). https://doi.org/10.1002/humu.21186 30. Wickenhagen WV, Salomons GS, Gibson KM, Jakobs C, Struys EA. Measurement of D-2-hydroxyglutarate dehydrogenase activity in cell homogenates derived from D-2-hydroxyglutaric aciduria patients. J Inherit Metab Dis 32, 264-268 (2009). https://doi.org/10.1007/s10545-009-1104-1 31. Losman JA, Kaelin WG, Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 27, 836-852 (2013). https://doi.org/10.1101/gad.217406.113 32. Tan AP, Mankad K. Intraventricular Glioblastoma Multiforme in A Child with L2-Hydroxyglutaric Aciduria. World neurosurgery 110, 288-290 (2018). https://doi.org/10.1016/j.wneu.2017.11.106 33. de Souza CF, et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep 23, 637-651 (2018). https://doi.org/10.1016/j.celrep.2018.03.107 34. Ceccarelli M, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 164, 550-563 (2016). https://doi.org/10.1016/j.cell.2015.12.028 35. Wilcken B, Pitt J, Heath D, Walsh P, Wilson G, Buchanan N. L-2-hydroxyglutaric aciduria: three Australian cases. Journal of inherited metabolic disease 16, 501-504 (1993). https://doi.org/10.1007/BF00711665 36. Barbot C, et al. L-2-Hydroxyglutaric aciduria: clinical, biochemical and magnetic resonance imaging in six Portuguese pediatric patients. Brain & development 19, 268-273 (1997). https://doi.org/10.1016/s0387-7604(97)00574-3 37. Wanders RJ, et al. L-2-Hydroxyglutaric aciduria: normal L-2-hydroxyglutarate dehydrogenase activity in liver from two new patients. Journal of inherited metabolic disease 20, 725-726 (1997). https://doi.org/10.1023/a:1005355316599 38. Ozisik PA, Akalan N, Palaoglu S, Topcu M. Medulloblastoma in a child with the metabolic disease L-2-hydroxyglutaric aciduria. Pediatric neurosurgery 37, 22-26 (2002). https://doi.org/10.1159/000065097 39. Topcu M, et al. L-2-hydroxyglutaric aciduria: a report of 29 patients. The Turkish journal of pediatrics 47, 1-7 (2005). 40. Vilarinho L, et al. Novel L2HGDH mutations in 21 patients with L-2-hydroxyglutaric aciduria of Portuguese origin. Human mutation 26, 395-396 (2005). https://doi.org/10.1002/humu.9373 41. Haliloglu G, et al. L-2-hydroxyglutaric aciduria and brain tumors in children with mutations in the L2HGDH gene: neuroimaging findings. Neuropediatrics 39, 119-122 (2008). https://doi.org/10.1055/s-2008-1081217 42. Aghili M, Zahedi F, Rafiee E. Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. Journal of neuro-oncology 91, 233-236 (2009). https://doi.org/10.1007/s11060-008-9706-2 43. İbrahim İnan BA, Ahmet Aslan, Begümhan Baysal, Ali Yıkılmaz. L-2 Hydroxyglutaric Aciduria and Anaplastic Oligodendroglioma: A Rare Association. Istanbul Medical Journal 19, 52-55 (2018). https://doi.org/10.1212/01.wnl.0000125335.21381.87 |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||