|
|
|
Free Neuropathology 4:24 (2023) |
|
Letter |
|
Adrenomyeloneuropathy (AMN): myelinopathy or axonopathy? Comment on: Considering the myelin-centric hypothesis: insights from Budka's historical adrenomyeloneuropathy case report by E. Salsano and C. Benzoni |
|
Herbert Budka |
|
Division of Neuropathology and Neurochemistry (Obersteiner Institute, formerly Institute of Neurology / Neurological Institute), Department of Neurology, Medical University of Vienna, Austria |
|
Corresponding author: |
|
Submitted: 13 December 2023 |
|
Keywords: Adrenomyeloneuropathy (AMN), Axonopathy, Axonopathy, Myelinopathy, Demyelination |
|
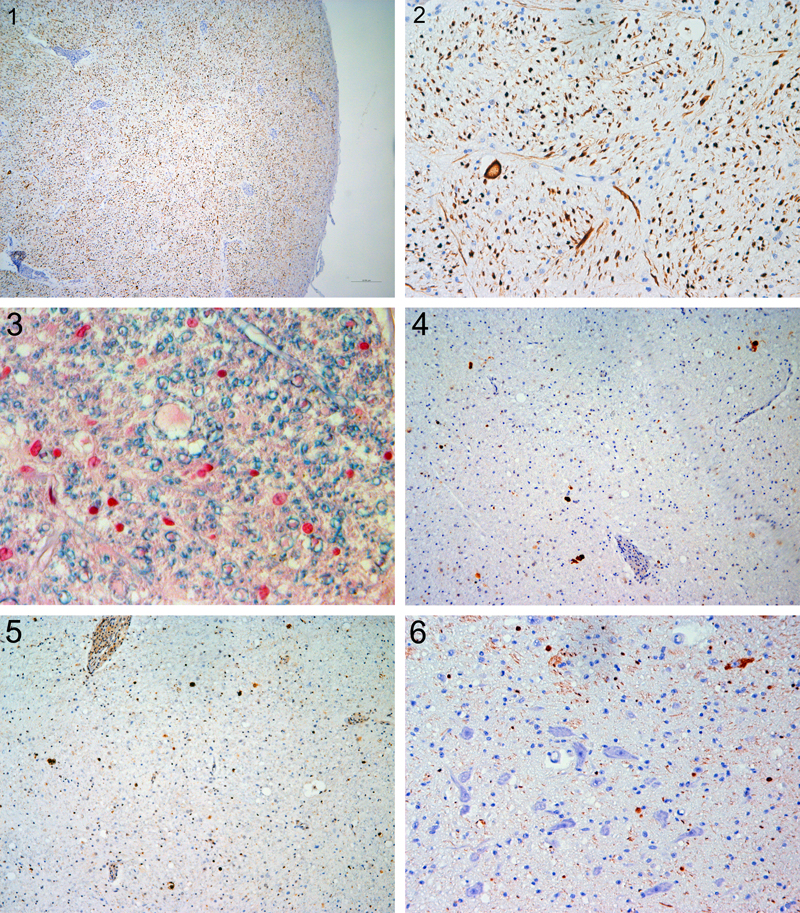
I am grateful to Drs. Salsano and Benzoni [1] for their appreciation of our original adrenomyeloneuropathy (AMN) report [2] and of my recent account on its history [3], but still more for raising an interesting scientific issue that, in my opinion, has not been completely resolved: whether the characteristic affection of long cerebrospinal tracts in AMN develops as either original myelin or axonal pathology, or even as both. They think I overlooked the issue. In fact, I have been well aware of it but decided – maybe wrongly – to omit such a discussion from my recent contribution that I considered as a primarily historic account in the sense of a "nice story", as it does not provide new scientific data [3]. However, I am now glad that Drs. Salsano and Benzoni’s letter offers the opportunity to comment on myelinopathy versus axonopathy in AMN. I agree with them that this pathogenetic issue is not purely academic: with future progress in molecular and personalized medicine, it becomes relevant to tailor therapy to the right target. In a potentially confusing way, the terminology of "demyelination" has been in twofold use. First, more generally and somehow imprecisely, it has been applied to plain myelin loss, including descriptions of histological stainings for myelin, regardless of the underlying pathogenesis. Second and more specifically, it has signified dismantling of the myelin sheath from the (at least initially) intact axon, resulting in myelino-axonal dissociation, as may be seen in oligodendrogliopathies (best example: the lytic infection by the John Cunningham virus/JCV in progressive multifocal leukoencephalopathy/PML) and Schwannopathies; cellular or humoral immune attacks against myelin, ideally to be visualized by myelin "stripping" by macrophages or binding of antibodies; or electrolytic or metabolic myelin impairment e.g. in central pontine myelinolysis/CPM or leukodystrophies. Indeed, X-linked or classic adrenoleukodystrophy (ALD) has been categorized with other leukodystrophies among demyelinating diseases. However, specific demyelination and axon injury are often concurrent and can have common pathogenic mechanisms and physiological effects [4]. Thus the situation is complicated by the mutual dependency between myelin sheath and axon, leading to secondary axonopathies in primary myelinopathies, and vice versa. So it might resemble the classical chicken-or-egg dilemma. In addition, the dissection of such pathogenetic events requires specific methodologies that can be met best in experimental models, but only to a limited degree in human autopsy tissue that provides a glimpse at one specific point in time, not over a period, and may be prone to autolytic and preparatory constraints as well as subjective interpretation. In our original AMN report [2], we described the lesioning of long tracts as "incomplete demyelination", meant in a more general sense and not considered to imply a specific pathogenesis that was then well beyond our original intent. We mainly emphasized perivascular histiocytic cuffs with pathognomonic ultrastructural inclusions that guided us towards ALD. However, we mentioned "well preserved axons" and "no loss of oligodendrocytes", based on then available basic histological stains without immunohistochemistry. Now, with hindsight by my 52 years of neuropathological experience and the stunning progress of biomedical sciences including neuropathology, I must criticize these imprecise descriptions of my early career. Indeed, when I made preparations for my recent historical account [3], original paraffin blocks were retrieved and immunostained with up-to-date methodology and antibodies, including axonal markers (phosphorylated and non-phosphorylated neurofilament proteins/NFP), ubiquitin and the autophagosome cargo protein p62, tau, and phosphorylated and non-phosphorylated TDP-43. While it is not easy to recognize relevant features in the original stainings of histological sections from our case that are now available to the public by virtual microscopy [3], re-examination with modern molecular markers gives a more clear-cut feeling of what happened in the affected tissue. First, affected cerebrospinal tracts show not only diffuse loss of myelin, resulting from moderate loss of myelinated fibers, but also moderate diffuse loss of pNFP-immunoreactive axons (Fig. 1) that seems to be commensurate with myelin loss. Second, there are some disseminated dystrophic axonal swellings (Fig. 2) that are occasionally found to be surrounded by a still intact myelin sheath (Fig. 3), and show immunoreactivity for p62 (Fig. 4) and ubiquitin (Fig. 5). Such axonal swellings are, to a lesser degree, disseminated also in the tegmentum. Third, rare tegmental neurons express pNFP in their cytoplasm (Fig. 6), suggesting a proximal "axonal reaction" to distal degeneration, whereas other neurons do not have pNFP in their cell bodies, the normal finding. Inclusion bodies or deposits of other proteins as examined were absent. In sum, these neuropathological features document an ongoing axonopathy with dystrophic features in long nerve fiber tracts, whereas evidence for a primary myelin affection is lacking, albeit difficult to appreciate in such tissue. In the literature on AMN, the pathology observed in the affected tracts has been usually described to reflect a distal axonopathy [5,6]. My re-examination of the original AMN case is in accordance with this interpretation. On the other hand, Griffin et al. [7] described, in two sural nerve biopsies of their five first serial AMN cases, onion bulbs, a feature diagnostic of primary demyelination in the peripheral nerve. In their letter, Drs. Salsano and Benzoni suggest a "hypothesis of a myelin-centric nature of early-stage AMN", and provide arguments for this viewpoint, mainly based on in vivo quantitative MRI techniques, and rescue from disease by human ABCD1 expression in oligodendrocytes of an ALD model in zebrafish [1]. In addition, they interpret neuropathological findings in our original case with a relatively short clinical duration to support their myelin-centric hypothesis. I apologize to Drs. Salsano and Benzoni that our imperfect description in the original report might have been misleading. However, I agree with them that more evidence on the fate of the myelin-axon axis in AMN is needed, including their proposal to study a mouse model with conditional knock-out of abcd1 in distinct CNS cell types. Acknowledgement I am greatly indebted to Prof. Romana Höftberger, PD Ellen Gelpi, and the Histo Lab Members of the Obersteiner Institute Vienna, for their support in re-examining the original AMN case.
All figures are taken from the pyramid or tegmentum of the lower medulla oblongata in new immunohistochemical sections (IHC, performed in a DAKO Autostainer with DAKO Envision Kit and diaminobenzidine/DAB as chromogen) or Luxol Fast Blue (LFB) stained sections of the original AMN case. Note also the characteristic perivascular histiocytic sleeves.
Fig. 1. Moderate diffuse loss of axonal profiles. IHC with anti-pNFPs (SMI31) x 40. References 1. Salsano E, Benzoni C: Considering the myelin-centric hypothesis: insights from Budka's historical adrenomyeloneuropathy case report. Free Neuropathol. 4: 23 (2023). https://doi.org/10.17879/freeneuropathology-2023-5218 2. Budka H, Sluga E, Heiss WD: Spastic paraplegia associated with Addison’s disease: adult variant of adreno-leukodystrophy. J Neurol. 213: 237–250 (1976). https://doi.org/10.1007/BF00312873 3. Budka H: A historical look using virtual microscopy: the first case report of adrenomyeloneuropathy (AMN). Free Neuropathol. 4: 18 (2023). https://doi.org/10.17879/freeneuropathology-2023-5115 4. Moore GRW, Stadelmann-Nessler C: Demyelinating Diseases. In: Love S, Budka, H, Ironside JW, Perry A (eds.) Greenfield’s Neuropathology, 9th ed., vol. 2, pp. 1297-1412. CRC Press, Boca Raton FL 2015. 5. Faust PL, Powers JM: Peroxisomal disorders. In: Love S, Budka, H, Ironside JW, Perry A (eds.) Greenfield’s Neuropathology, 9th ed., vol. 1, pp. 562-588. CRC Press, Boca Raton FL 2015. 6. Schaumburg HH, Powers JM, Raine CS, Spencer PS, Griffin JW, Prineas JW, Boehme DM: Adrenomyeloneuropathy: a probable variant of adrenoleukodystrophy. II. General pathologic, neuropathologic, and biochemical aspects. Neurology 27 (12): 1114-1119 (1977). https://doi.org/10.1212/wnl.27.12.1114 7. Griffin JW, Goren E, Schaumburg H, Engel WK, Loriaux L: Adrenomyeloneuropathy: A probable variant of adrenoleukodystrophy. I. Clinical and Endocrinologic Aspects. Neurology 27 (12): 1107–1113 (1977). https://doi.org/10.1212/WNL.27.12.1107
Copyright: © 2023 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |