|
|
|||||||||
|
Free Neuropathology 4:18 (2023) |
|||||||||
|
Flashback |
|||||||||
|
A historical look using virtual microscopy: the first case report of adrenomyeloneuropathy (AMN) |
|||||||||
|
Herbert Budka |
|||||||||
|
Division of Neuropathology and Neurochemistry (Obersteiner Institute, formerly Institute of Neurology / Neurological Institute), Department of Neurology, Medical University of Vienna, Austria |
|||||||||
|
Corresponding author: |
|||||||||
|
Submitted: 11 September 2023 |
|||||||||
|
Keywords: Adrenoleukodystrophy (ALD), Adrenomyeloneuropathy (AMN), Peroxisomal diseases, Peroxisomes, Neuropathology, History |
|||||||||
|
Abstract The history of adrenoleukodystrophy (ALD), adrenomyeloneuropathy (AMN) and other peroxisomal diseases is exemplary for the stunning progress of scientific medicine within the past 50 years. Like many breakthroughs in medicine, the detailed analysis of patients’ pathologically affected tissues was instrumental, resulting in stepwise systematic clarification of what had remained enigmatic until the 1970s. This flashback paper is a recollection of the first neuropathological description of a slowly evolving clinical phenotype, spastic paraparesis with adrenal insufficiency, in a young adult by Budka et al. 1976 [3], using virtual microscopy of the original histologic slides. The clinico-pathological presentation derives from the classical cerebral ALD phenotype in boys, where electron microscopy demonstrated the underlying pathological hallmark of characteristic lipid inclusions shared by both phenotypes. Our report allowed the delineation of a new disease type almost simultaneously described in more cases as AMN by Griffin et al. 1977 [4] and Schaumburg et al. 1977 [11]. Moreover, our report indicated clinical heterogeneity in the ALD disease group that, as shown later, extends further to females, to Addison-only, and even to asymptomatic subjects. The gene underlying ALD was discovered in 1993 as a defect in the ABCD1 gene. Yet, it has hitherto remained unclear how the gene defect causes the strikingly broad and unpredictable phenotypic spectrum of ALD/AMN. |
|||||||||
|
The history of what is now established as adrenoleukodystrophy (ALD), adrenomyeloneuro-pathy (AMN) and other peroxisomal diseases is exemplary for the stunning progress of scientific medicine/medical science within the past 50 years. This journey starts with the mere description of clinico-pathological phenotypes, follows up with the identification of a biochemical abnormality, and culminates by defining the molecular and genetic basis of the disease, by newborn screening, and by promising therapies, as recently reviewed [17]. Instrumental, like in many breakthroughs in medicine, was the detailed analysis of patients’ pathologically affected tissues, resulting in step-wise systematic clarification of what had remained an obscure enigma until the 1970s. Moreover, the history of ALD and AMN is exemplary for the multidisciplinary and international character of contributing studies as a prerequisite for success. This article recollects an early step in our knowledge of the disease group where neuropathology had a pivotal role. Local background of the first case of AMN The present (hi)story evolved more than 50 years ago, after I became a trainee at the renowned Neurological Institute, or Obersteiner Institute, in Vienna from late 1971, right after my graduation as an MD. Kurt Jellinger (born 1931) was there in charge of clinical neuropathology, made the preliminary neuropathological autopsy report of an interesting patient who died in late 1972, and suggested me to write up the story. However, I was extremely busy in learning all elements of classical neuropathology and doing clinical training in parallel to become specialist in neurology and psychiatry. Moreover, I was not yet aware of the paramount importance of publishing in science. So I failed to give priority to this task. In addition, my early years were increasingly overshadowed by Kurt’s leave from the Institute in 1975, a severe blow to diagnostic neuropathology and a disaster for me, as I was suddenly expected to bear most of the diagnostic workload myself; I gave a glimpse on those difficult early years elsewhere [2]. So it was May 1976 when the article was finally submitted, and was printed in the September issue of the Journal of Neurology (formerly: Deutsche Zeitschrift für Nervenheilkunde and Zeitschrift für Neurologie) [3] that then had a mixture of German- and English-written articles. My co-authors were Elfriede Sluga (1930-2008, Fig. 1) and Wolf-Dieter Heiss (born 1939, Fig. 2), both much more senior than me (Fig. 3).
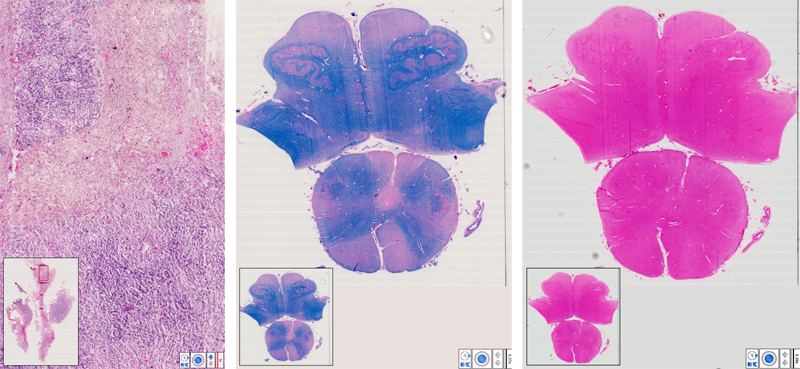
Elfriede (“Fritzi”) had pioneered the diagnostic use of the electron microscope (EM) at the Neurological Institute, mostly for neuromuscular biopsies, had a dominant clinical interest and later became professor and director of a department of neurology at a municipal hospital in Vienna, in Kurt Jellinger’s footsteps who had done the same earlier. By then, I never had a chance to touch the EM, so Fritzi contributed the essential EM diagnostics of the report. Wolf-Dieter was then a consultant neurologist at the Neurological University Clinic in Vienna, responsible for a ward of some 20 hospital beds where the patient was admitted, and an impressive teacher during my clinical training. He oversaw clinical details of the report and later became professor at the University of Cologne and director of the Max Planck Institute for Brain Research in Köln-Merheim, and is now widely recognized as a top expert on cerebral hemodynamics, including a pioneering role in the establishment of stroke units. Wolf-Dieter, a giant in clinical neurosciences in his own right, originally went to Cologne to succeed another giant, Klaus-Joachim Zülch (1910-1988) who had combined clinical neurology with neuropathology as neuro-oncologist and stroke expert; he is now remembered as main author of the very first edition of the famous WHO Blue Book Series on Histological Typing of Tumours of the CNS [18]. For the Neurological Institute in Vienna, our report laid the foundation for a new research and diagnostic area that became important not only for the Institute’s neuropathology branch, but still more so for its Division of Neurochemistry, led by Hans (“Hanno”) Bernheimer (1930-2016), and mainly involving Brunhilde Molzer (born 1940) [7] with very long chain fatty acids (VLCFA) diagnostic biochemistry for ALD/AMN suspects. In the 1990s Johannes Berger (born 1964) joined the neurochemistry group and expanded its molecular and experimental studies; from 2000, he has done research on peroxisomes as Professor in the newly founded Center of Brain Research in Vienna. The patient Our propositus was a relatively healthy – well, not quite – man who succumbed quickly and rather unexpectedly in 1972 at the age of 24, after a slowly evolving disease with spastic paraparesis over 2 years. No definite diagnosis was made during lifetime. You can make yourself familiar with the (neuro)pathology at autopsy by viewing scanned slides of the right adrenal gland (Fig. 4) and of the medulla oblongata (Figs. 5 & 6). Indeed, I strongly recommend you to stop here and do your virtual microscoping before you continue to read more details. There are of course different types of observers, but my favorite personal style has always been the detective-style approach, starting with a few basic data (sufficient here in the preceding paragraph), and aiming to have a more complete story evolving from the discoveries during microscoping (and earlier autopsy with brain cutting). If you read the more detailed description already at start, you might easily and quickly look at everything, but it may happen that you do not SEE in Thoreau’s sense (see citation at the start of this article). Moreover, the suggested early microscoping reflects the usual neuropathologist’s way of working: ultimately, you learn most from what you initially might NOT see. Recapitulation of the case report In our report, we made a detailed description of the clinical course, laboratory findings, and results of a complete autopsy including histology and electron microscopy of various organs. There is nothing new that can be added since then. So an excerpt follows for those who are unable or unwilling to read the original paper. Our propositus had a family history of an unusual dark skin pigmentation of his mother and was hyperpigmented since childhood. Originally, this was an active young man who used to make bicycle tours. At age 22, progressive weakness was noted in both legs. Subsequently, bladder and anal sphincter dysfunction developed; sexual functions were unimpaired. Seven months before death, he presented at the Neurologic Clinic in Vienna with a deeply bronzed skin, alopecia including axillary and pubic hairs, and a spastic gait with bilaterally exaggerated tendon reflexes of legs, bilateral Babinski’s sign, and loss of abdominal reflexes. There was hypesthesia of both legs for all qualities, with distal accentuation. When Wolf-Dieter Heiss first saw the patient, he immediately suspected Addison’s disease, because of the bronzed skin. However, extensive involvement of endocrinology specialists did not result in such a clear-cut diagnosis during the lifetime of the patient. Thereafter, laboratory results as detailed in our report were interpreted as compatible with primary adrenal insufficiency. After a visit to a sauna bath 2 weeks before death, the patient deteriorated rapidly, with collapse, vomiting, vertigo, and impairment of consciousness alternating between agitation and sopor. One week before death, hyperthermia and progressive metabolic and circulatory dysregulation developed. Death was caused by cardiovascular failure. A complete autopsy at the Institute of Pathology in Vienna was performed by the then famous Prof. Lothar (“Rex”) Kucsko (1912-1976) who contributed also otherwise to my career [2]. He described the adrenals as atrophic, firm, chocolate-brown and without discernible structure, but including two small nodules on the right side; his final diagnosis was so-called immune adrenalitis, with an acute Addison crisis as cause of death. He was perfectly right on the latter, less so on the former. Histology of various other organs was characterized by widespread scattered lymphocytic infiltrates. Under the microscope: what you may see Fig. 4 Adrenal (HE). The organ structure is disorganized, with intact medulla (bluish stained tissue) but only focal remnants of cortical tissue (reddish stained tissue) with large, sometimes bizarre and ballooned cells with occasional cytoplasmic “striations”. In addition, there is prominent fibrosis and focal lymphocytic infiltration. Fig. 5 & 6 Medulla oblongata (LFB, HE). There is symmetric degeneration of pyramids, to a lesser degree of medial and lateral lemnisci, spinocerebellar and Goll’s tracts. In addition to perivascular lymphocytic infiltrates, there are perivascular cuffs of histiocytes with epitheloid appearance and occasional cytoplasmic “striation” (I am not sure how obvious the striation is on 2-D virtual files that don’t allow a 3-D impression, in contrast to old-fashion microscoping with a glass slide when you can play with the focusing screw).
Medico-scientific background For decades, what is now called ALD has been a rare and poorly understood, but rapidly evolving and fatal demyelinating disease of male kids, a combination of inflammatory diffuse sclerosis of the brain with adrenal insufficiency. Haberfeld and Spieler have been credited with the first clinico-pathological description of classical childhood ALD [5], followed by articles by Schilder who gave his name to what he called encephalitis periaxialis diffusa [13][14][15]. In the 1970s, most research on ALD was concentrated at the Albert Einstein College of Medicine in Bronx, New York. The Department of Pathology there was a hotbed of neuropathology, with eminent researchers like Robert D. Terry and Henryk M. Wisniewski who contributed landmark studies in Alzheimer disease, and John W. Prineas and Cedric Raine who did the same in multiple sclerosis. A young trainee, James M. Powers, teamed up with Herbert H. Schaumburg, an expert in neurotoxicology and peripheral nerves, Ann Johnson and Kunihiko Suzuki, to study a disease that just recently had been termed ALD [1]; their work over the next few years elucidated most of its (neuro)pathology and biochemistry. Powers and Schaumburg demonstrated characteristic lipid inclusions in ALD adrenals by EM examination [9], followed by their description in brain [12] and other organs [10]. Subsequent neurochemical studies showed a striking excess of VLCFA in brain cholesterol esters as substrate of ALD inclusions [6], clearing the path to biochemical assays that subsequently have dominated diagnostics in samples of cultured skin fibroblasts, amniocytes and plasma. Independently from our report, Griffin et al. [4], and Schaumburg et al. [11], published in 1977 a small clinico-pathologic series of patients with familial spastic paraparesis with Addison’s disease, ALD-like ultrastructure and biochemistry, and coined the term adrenomyeloneuropathy (AMN). From the 1980s up to now, much of clinical ALD/AMN research originated from the Kennedy-Krieger Research Institute in Baltimore, MD, led by Hugo Moser (1924-2007), the unrivalled eminence in peroxisomal diseases, and his wife Ann, resulting in establishment of methods for early diagnosis and culminating in identification of the genetic basis by defects of the ALDP/ABCD1 gene [8] encoding for a peroxisomal membrane protein and member of the ATP-binding cassette (ABC) transporter family. On top of that, Kennedy-Krieger contributed much more that is by now known in peroxisomal diseases. Moreover, they had a major role in development and assessment of therapies such as “Lorenzo’s Oil“ of Hollywood fame, bone marrow and hematopoietic stem cell transplantation, and gene therapy [17]. Still more impressively, Hugo and Ann Moser established an absolutely exemplary relation to their ALD/AMN patients and their families who returned their love and affection to these two outstanding advocates for persons suffering from neurological disabilities. AMN (neuro)pathology The (neuro)pathology of AMN is typical but not specific without appropriate clinical information including family history, VLCFA biochemistry and/or EM findings. Nowadays genetic testing for the ALDP/ABCD1 gene is the mainstay of definite diagnostics. Neuropathology, as seen in the virtual slides, is characterized by two hallmarks: first, degeneration of long tracts of brainstem and spinal cord in a pseudosystemic distribution, and second, the presence within degenerating long tracts of perivascular sleeves of lipid-filled histiocytes that rarely may, or may not, give a “striated” impression. This striation might be the light microscopical appearance of the circumscribed packaging of lamellar lipid aggregates with clear clefts as seen by EM. In parallel, such degenerative changes may also be present in the peripheral nervous system. The scattered lymphocytic infiltrates are to be interpreted as secondary to the adrenal insufficiency that may, or may not, be manifest in an otherwise typical AMN. The pathology of the adrenal glands reflects the degree of adrenal insufficiency, with storage of lipids in enlarged cortical cells. Originally, in addition to histology, EM examination was important to document peculiar lipidic inclusions with a lamellar “leaflet” structure in perivascular histiocytes of the oblongata pyramids, as described and illustrated on two pages of our report, and found earlier as characteristic of ALD by Powers and Schaumburg [9] and Schaumburg et al. [12]. We suggested the ultrastructural inclusions in the CNS to derive from degenerated myelin in the affected tracts, linking ALD/AMN to pathological storage of a myelin degradation product [3]. Significance Our study [3] allowed the delineation of what became a new disease type, a little bit later termed AMN [4][11]. The presentation deviated from the classical cerebral ALD phenotype in boys. However, histology and electron microscopy demonstrated the underlying pathological hallmark of characteristic inclusions to be shared by both phenotypes. Moreover, as the first pathological description of slowly evolving spastic paraparesis with adrenal insufficiency in young adults, it indicated clinical heterogeneity in the ALD disease group that, as shown later, extends further to females [7] with the same heterogeneity as in males, to Addison-only, and even to asymptomatics [17]. Neonatal ALD is now considered another disease with different genetic defects. Despite all progress, however, it has remained unclear how defects of the ABCD1 gene cause the strikingly broad, and unpredictable, phenotypic spectrum of ALD/AMN. Our publication has met moderate interest, at least in scientometric terms: by Sept. 3, 2023, Google Scholar listed 125 citations. Indeed, in early years after the publication citations were rather scarce, until Hugo Moser started to cite it in his reviews on the history of the field. It was a great pleasure and honor when he was visiting our Institute in Vienna a few times, and I was greatly impressed by his personality, openness and kindness, knowledge and willingness to co-operate on behalf of his patients. I was also happy to have met Jim Powers a few times, a kind and experienced colleague and now the foremost neuropathologist of ALD/AMN. We discussed our mutual interests, and it is clear that he – with Herb Schaumburg, Cedric Raine, Jack Griffin, Peter Spencer, John Prineas and others – published their AMN cases in the December 1977 issue of Neurology [11] without being aware of our preceding publication. Indeed there was no Pubmed or Internet at that time. Anyway, our stories are another example that scientific progress may occur simultaneously and independently in distant locations and settings. References 1. Blaw ME: Melanodermic type leukodystrophy (adreno-leukodystrophy). In: Vinken PJ, Bruyn GW (eds): Handbook of Clinical Neurology vol.10, pp. 128-133. Amsterdam: North Holland 1970 ISBN 9780444100597, 0444100598 OCLC 872408480 2. Budka H: Neuropathology through the ages – personal reflections: The golden era of neuropathology. Free Neuropathol 1: 15 (26 pp.) (2020). https://www.uni-muenster.de/Ejournals/index.php/fnp/article/view/2817 3. Budka H, Sluga E, Heiss W-D: Spastic paraplegia associated with Addison's disease: adult variant of adreno-leukodystrophy. J Neurol 213 (3): 237-250 (1976). https://doi.org/10.1007/BF00312873 4. Griffin JW, Goren E, Schaumburg H, Engel WK, Loriaux L: Adrenomyeloneuropathy: A probable variant of adrenoleukodystrophy. I. Clinical and Endocrinologic Aspects. Neurology 27 (12): 1107–1113 (1977). https://doi.org/10.1212/WNL.27.12.1107 5. Haberfeld W, Spieler F: Zur diffusen Hirn-Rückenmarksklerose im Kindesalter. Deutsche Z Nervenheilk 40: 436–463 (1910). https://doi.org/10.1007/BF01629013 6. Igarashi M, Schaumburg HH, Powers J, Kishmoto Y, Kolodny E, Suzuki K: Fatty acid abnormality in adrenoleukodystrophy. J Neurochem 26, 851–860 (1976). https://doi.org/10.1111/j.1471-4159.1976.tb04462.x 7. Molzer B, Bernheimer H, Budka H, Pilz P, Toifl K: Accumulation of very long chain fatty acids is common to 3 variants of adrenoleukodystrophy (ALD): "classical" ALD, atypical ALD (female patient) and adrenomyeloneuropathy. J Neurol Sci 51: 301-310 (1981). https://doi.org/10.1016/0022-510x(81)90108-8 8. Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, Moser H, Poustka A-M, Mandel J-L, Aubourg P: Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361: 726–730 (1993). https://doi.org/10.1038/361726a0 9. Powers JM, Schaumburg HH: The adrenal cortex in adreno-leukodystrophy. Arch Pathol 96: 305-310 (1973). PMID: 4741901 10. Powers JM, Schaumburg HH: Adrenoleukodystrophy (sex-linked Schilder’s disease). A pathogenetic hypothesis based on ultrastructural lesions in adrenal cortex, peripheral nerve and testis. Am J Pathol 76: 481-500 (1974). PMID: 4741901 11. Schaumburg HH, Powers JM, Raine CS, Spencer PS, Griffin JW, Prineas JW, Boehme DM: Adrenomyeloneuropathy: a probable variant of adrenoleukodystrophy. II. General pathologic, neuropathologic, and biochemical aspects. Neurology 27 (12): 1114-1119 (1977). https://doi.org/10.1212/wnl.27.12.1114 12. Schaumburg HH, Powers JM, Suzuki K, Raine CS: Adrenoleukodystrophy (sex-linked Schilder disease). Ultrastructural demonstration of specific cytoplasmic inclusions in the central nervous system. Arch Neurol 31: 210-213 (1974). https://doi.org/10.1001/archneur.1974.00490390092013 13. Schilder PF: Zur Kenntnis der sogenannten diffusen Sklerose (über Encephalitis periaxialis diffusa). Z ges Neurol Psychiat 10, 1–60 (1912). https://doi.org/10.1007/BF02901445 14. Schilder PF: Zur Frage der Encephalitis periaxiallis diffusa (sogenannte diffuse Sklerose). Z ges Neurol Psychiat 15, 359–376 (1913) 15. Schilder PF: Die Encephalitis periaxiallis diffusa (nebst Bemerkungen ü̈ber die Apraxie des Lidschlusses). Arch Psychiat 71, 327–356 (1924) 16. Thoreau HD: Quotes. Retrieved September 3, 2023, from BrainyQuote.com https://www.brainyquote.com/search_results?x=0&y=0&q=Thoreau 17. Turk BR, Theda C, Fatemi A, Moser AB: X-linked adrenoleukodystrophy: pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Internat J Develop Neurosci 80: 52-72 (2020). https://doi.org/10.1002/jdn.10003 18. Zülch KJ: Histological Typing of Tumours of the Central Nervous System. International Histologic Classification of Tumours, No. 21. WHO: Geneva 1979, ISBN 9789241760218, 9241760214 OCLC 567810677
Copyright: © 2023 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |
|||||||||