|
|
|
Free Neuropathology 4:13 (2023) |
|
Review |
|
Neurodegeneration: 2023 update |
|
John F. Crary |
|
Department of Pathology, Nash Family Department of Neuroscience, Department of Artificial Intelligence & Human Health, Neuropathology Brain Bank & Research CoRE, Ronald M. Loeb Center for Alzheimer's Disease, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA |
|
Corresponding author: |
|
Submitted: 06 June 2023 |
|
Keywords: Neurodegeneration, Neuropathology, Aging, Alzheimer disease, Tauopathy, α-synucleinopathy, TDP-43 proteinopathy, Traumatic brain injury |
|
Abstract This paper reviews ten highly impactful studies published in the previous year selected by the author from the neurodegenerative neuropathology literature. As in previous years, the focus is to highlight human tissue-based experimentation most relevant to neuropathologists. A concerted effort was made to balance the selected studies across disease categories, approaches, and methodologies to capture the breadth of the research landscape. Studies include an integrated proteomic and transcriptomic study of Alzheimer disease (AD) and new consensus diagnostic neuropathological criteria for progressive supranuclear palsy. A number of studies looking at TAR DNA-binding protein 43 (TDP-43) are highlighted. One examined interaction between AD and limbic age-related TDP-43 encephalopathy (LATE) and yet another demonstrated how TDP-43 represses cryptic exon inclusion in UNC13A, suggesting a novel pathogenic mechanism. Most surprisingly, three cryogenic electron microscopy (cryo-EM) studies showed that TMEM106B filaments form the core of TDP-43-positive inclusions. Cryo-EM revealed a prion protein amyloid structure from aggregates in Gerstmann-Sträussler-Scheinker disease. There was an elegant functional genomic study cataloging microglial gene expression in the human brain. A study shed light on how APOE influences chronic traumatic encephalopathy. A pathoanatomical study tested the dual hit hypothesis of Lewy body progression throughout the nervous system. And finally, deep learning continues to show its promise with application of a weakly supervised multiple instance learning paradigm to assess aging post-mortem brains. |
|
1. An integrated proteomic and transcriptomic analysis of Alzheimer disease reveals unique pathways beyond mRNA Powerful transcriptomic approaches are increasingly being applied to post-mortem Alzheimer disease (AD) tissues and other diseases. Historically, methods included hybridization-based microarrays and bulk RNA-sequencing, but increasingly single cell RNA profiling and spatial transcriptomics are becoming routine. Proteins, owing to their increased complexity, are more challenging to assess, but are ostensibly more proximate to the relevant alterations that drive neurodegeneration. A study by Johnson et al. entitled “Large-scale deep multi-layer analysis of Alzheimer's disease brain reveals strong proteomic disease-related changes not observed at the RNA level” analyzed proteomes of more than 1,000 brain tissues with a goal of trying to better understand the biological processes disrupted in AD (Johnson et al., 2022). The investigators deployed tandem mass tag mass spectrometry (TMT-MS), an approach used to quantitatively analyze and compare protein expression levels that uses isobaric tagging to enable multiplexed quantification of peptides and proteins. TMT-MS offers several advantages, including high multiplexing capacity, accurate and reproducible quantification, and the ability to analyze complex proteomes. Using this methodology, the research team identified new AD-related protein co-expression modules that were highly preserved across cohorts and brain regions. Remarkably, nearly half of the protein co-expression modules were not observed in mRNA networks. Two notable AD-associated modules that were unique to the proteomic network were related to mitogen-activated protein kinase (MAPK) signaling and the matrisome (Figure 1). The matrisome refers to the collection of extracellular matrix (ECM) proteins and associated factors and is a complex network of proteins and other molecules that provide structural support and regulate various cellular processes. The MAPK/metabolism module was associated with the rate of cognitive decline whereas the matrisome module was influenced by the APOE ε4 allele. These results indicate that pathways that are unique to the proteome could lead to therapeutic targets and biomarkers.
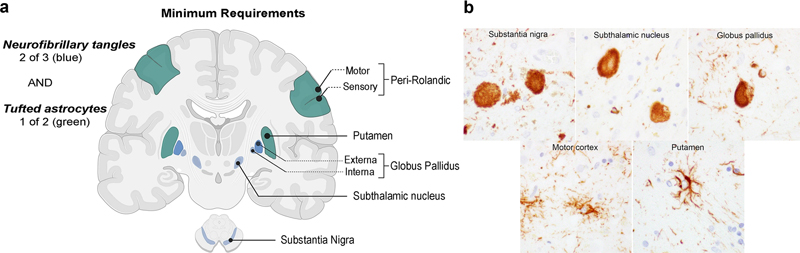
Figure 1. Tandem mass tag mass spectrometry (TMT-MS) derived Alzheimer disease protein network contains modules that are not present in transcriptomic analysis. a. Experimental overview. b. Modules that had a Zsummary score greater than or equal to 1.96 (or q = 0.05, blue dotted line) were considered to be preserved. Unique modules included MAPK signaling and the matrisome (reproduced from Johnson et al., 2022 under the creative commons license). 2. Revision of the neuropathological criteria for progressive supranuclear palsy The National Institute of Neurological Disorders and Stroke previously advanced neuropathologic criteria for progressive supranuclear palsy (PSP), which have been the standard for nearly 30 years. However, the complex evaluation process and dependence on non-uniform staining methods limited the utility of the criteria. Since then, advancements in immunohistochemistry and the availability of specific antibodies targeting pathological tau proteins, especially phosphorylation-dependent epitopes, have improved the recognition of key PSP neuropathological features, especially tufted astrocytes. Further, new diagnostic entities have emerged, including chronic traumatic encephalopathy (CTE), aging-related tau astrogliopathy (ARTAG) and globular glial tauopathy GGT, which required better delineation from PSP. To update the neuropathologic criteria for PSP, an international team of neuropathologists, supported by the Rainwater Charitable Foundation / Tau Consortium proposed criteria that incorporate recent advancements in tauopathies and are easier to apply in non-specialized clinical settings (Roemer et al., 2022). The goal was to develop criteria with high sensitivity, specificity, and inter-rater agreement, even for atypical variants of PSP, while being independent of clinical information and suitable for non-specialized neuropathology services. The investigators evaluated digital slides from 10 brain regions stained with hematoxylin and eosin and phosphorylated tau (AT8) immunohistochemistry for 15 typical and atypical PSP cases and 10 other tauopathies. Blinded to clinical and neuropathological information, 14 expert neuropathologists provided a categorical diagnosis (PSP or not-PSP) based on provisional criteria requiring neurofibrillary tangles or pretangles in two of three regions and tufted astrocytes in one of two regions. The provisional criteria had high sensitivity (0.97) and specificity (0.91), as well as almost perfect inter-rater reliability for diagnosing PSP and differentiating it from other tauopathies. The Rainwater Charitable Foundation criteria for the neuropathologic diagnosis of PSP feature a simplified diagnostic algorithm based on phosphorylated tau immunohistochemistry and incorporate tufted astrocytes as an essential diagnostic feature (Figure 2).
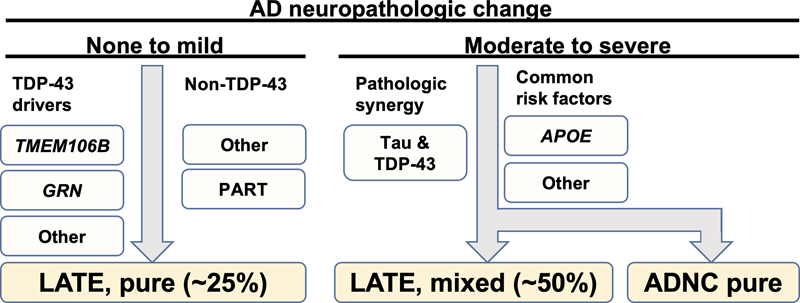
Figure 2. Rainwater neuropathological criteria for progressive supranuclear palsy. a. The minimum requirements are demonstration of neurofibrillary tangles in two any of three classically involved regions (e.g., the subthalamic nucleus, globus pallidus, and substantia nigra) alongside tufted astrocytes in the putamen or peri-Rolandic cortex. b. Examples of neurofibrillary tangles and tufted astrocytes on phospho-tau (AT8 antisera) immunohistochemical staining (reproduced with permission from Roemer et al., 2022 under the creative commons license). 3. Limbic-predominant age-related TDP-43 encephalopathy and Alzheimer disease: the impact of coexistence on cognition Limbic-predominant age-related TDP-43 encephalopathy neuropathologic change (LATE-NC) and Alzheimer disease neuropathologic change (ADNC) are both implicated in cognitive impairment among older individuals. However, the frequency of LATE-NC across the entire range of ADNC remains uncertain. To address this gap in knowledge, Nelson et al. performed a comprehensive analysis from 13 high-quality longitudinal studies, encompassing a total of 6,196 participants from diverse countries (Nelson et al., 2022). Among individuals with documented cognitive status prior to death, 43.0% were found to be cognitively normal, 14.9% had mild cognitive impairment, and 42.4% had dementia, consistent with epidemiological data for this age range. In cases with available CERAD neuritic amyloid plaque score data, 39.4% were confirmed to have LATE-NC at any stage based on autopsy findings. Notably, among brains with "frequent" neuritic amyloid plaques, 54.9% exhibited concurrent LATE-NC, while 27.0% of brains lacking detectable neuritic amyloid plaques displayed LATE-NC. Remarkably, LATE-NC was present in nearly 40% of participants and often coexisted with ADNC. Detailed neurocognitive assessments conducted proximate to death across 10 cohorts demonstrated a tendency for worse cognition in individuals with LATE-NC across the entire range of ADNC severity. None of the participants received a clinical diagnosis of definite frontotemporal dementia or a pathological diagnosis of frontotemporal lobar degeneration with TDP-43 inclusions. These findings provide valuable insights into the prevalence and interplay between LATE-NC and ADNC, highlighting their impact on cognitive function in the aging population.
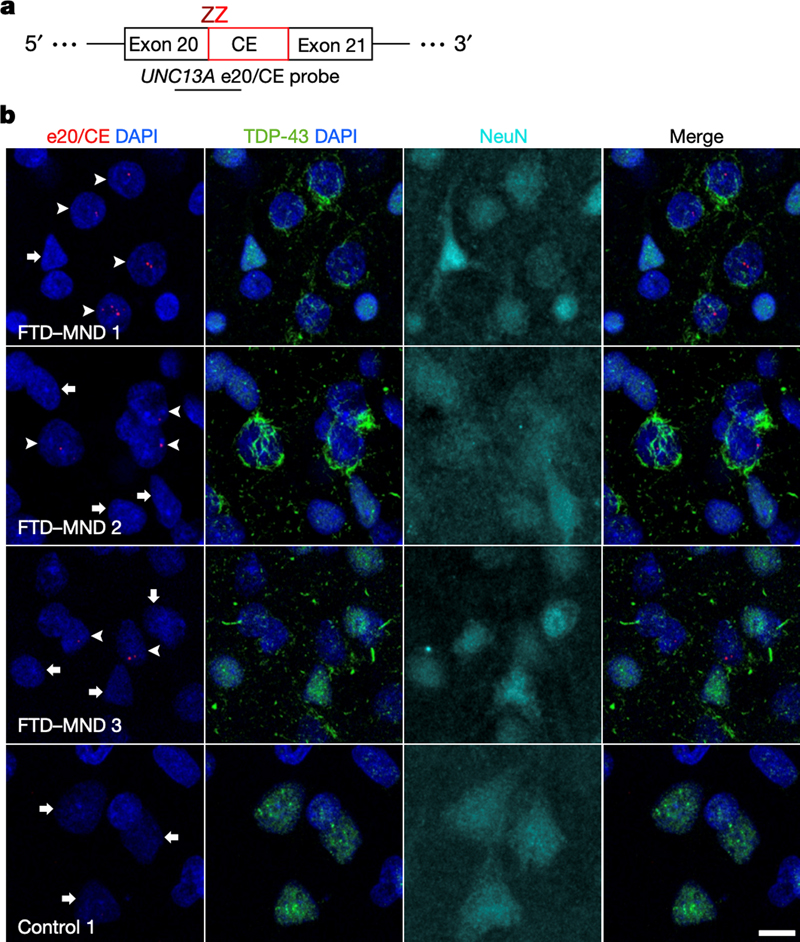
Figure 3. Frequency of limbic-predominant age-related TDP-43 encephalopathy neuropathologic change (LATE-NC) across the entire range of Alzheimer disease neuropathologic change (ADNC). In participants with minimal ADNC, ~25% have LATE-NC, suggesting ADNC-independent mechanisms. LATE-NC is associated with more severe primary age-related tauopathy pathology, indicating potential synergy. In subjects with severe ADNC, approximately 50% of participants had LATE-NC that may be driven by genetic factors (e.g., APOE genotype) or other factors. 4. TDP-43 represses cryptic exon inclusion in UNC13A Most amyotrophic lateral sclerosis (ALS) and a subset of frontotemporal lobar degeneration cases display TAR DNA-binding protein 43 (TDP-43) pathology. This is neuropathologically reflected by translocation of phosphorylated forms of the protein from the nucleus to the cytoplasm, a phenomenon that is readily visible on immunohistochemical stains. The significance and functional implications of this translocation are not clear, but may result in loss of TDP-43 function in the nucleus. TDP-43 is an RNA-binding protein with numerous roles, including repression of the inclusion of cryptic exons during splicing of heterogeneous RNA while in the nucleus. Previous studies have highlighted several genes, including POLDIP3 and STMN2 that exhibit aberrant splicing following TDP-43 depletion. In a paper by Ma et al., investigators sought to identify additional genes that exhibit cryptic splicing changes that are regulated by TDP-43 (Ma et al., 2022). Using post-mortem brain tissues from patients with Frontotemporal lobar degeneration (FTLD) and ALS, the authors used fluorescence-activated cell sorting to isolate nuclei with nuclear TDP-43 protein expression from nuclei lacking TDP-43 and performed RNA-seq. They found 66 genes with altered splicing using this analysis, including UNC13A, which had previously been identified in genome wide association studies with unclear mechanisms of increased risk. Specifically, they found a cryptic exon that is retained in cells with TDP-43 pathology that could be detected with exon splice site specific probes (Figure 4). Further cellular and histological validation supported these findings. It is known from studies in C. elegans and other systems that UNC13A encodes for a synaptic multidomain protein that participates in vesicle priming prior to fusion. These findings provide a direct mechanistic link between loss of TDP-43 function and a strong genetic risk factor for ALS-FTD.
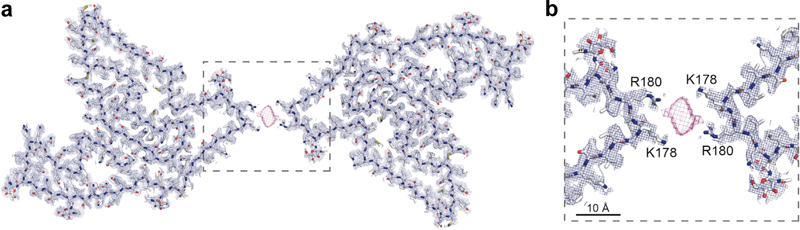
Figure 4. UNC13A cryptic splicing is associated with loss of nuclear TDP-43 in patients with FTD and motor neuron disease. a. BaseScope probe design b. in situ hybridization using the UNC13A e20/CE probe, combined with immunofluorescence for TDP-43 and NeuN illustrating the presence of UNC13A CE (arrowheads) in neurons showing depletion of nuclear TDP-43. Scale bar, 10 μm. (Reproduced from Ma et al., 2022 under the creative commons license). 5. A surprising turn in frontotemporal dementia FTLD is the third most common neurodegenerative disorder after Alzheimer’s and Parkinson’s disease, with one major FTLD subtype characterized by pathological neuronal inclusions with TDP-43 immunoreactivity. Determining the atomic structure of these inclusions is a high research priority given the expected utility for facilitating biomarker and therapeutic discovery. Using cryogenic electron microscopy (cryo-EM), three independent groups addressed this gap in knowledge with all making an astonishing discovery: the extracted amyloid fibrils were composed of fragments of transmembrane protein 106B (TMEM106B), a lysosomal membrane protein previously implicated as a genetic risk factor for FTLD-TDP (Chang et al., 2022; Jiang et al., 2022; Schweighauser et al., 2022). While details among the studies differ, in essence the inclusions consist of a normally intraluminal 135-residue fragment of TMEM106B that is likely shed before assuming this abnormal configuration. The aggregates, which are derived from the C-terminus, were likely missed in previous studies because most antisera target the N-terminus of the molecule. These findings raise a flood of new questions about TMEM106B and abnormal lysosomal activity in neurodegeneration.
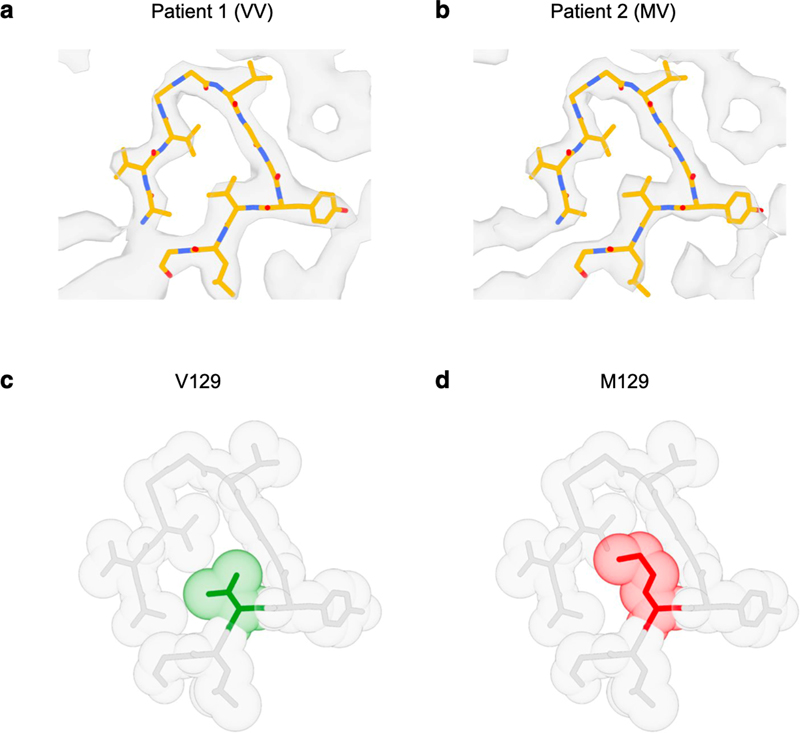
Figure 5. TMEM106B cryo-EM doublet fibril. (Reproduced with permission from Chang et al., 2022 under the creative commons license). 6. The cryo-EM structure of prion protein amyloid There are a number of dominantly inherited prion protein (PrP) amyloidoses, including Gerstmann-Sträussler-Scheinker disease (GSS), PrP cerebral amyloid angiopathy, and prion amyloidosis with variable phenotypes. All are caused by mutations in the prion protein gene (PRNP). GSS is notable in that typical extracellular deposits made of PrP amyloid (APrP) are associated with abnormal intraneuronal tau inclusions that are identical to those observed in classical AD, which raises fundamental questions as to how amyloid-beta causes tau pathology and whether there are shared pathogenic mechanisms. Understanding the atomic structure of the APrP has the potential to shed light on how extracellular amyloid deposition in general might trigger tau pathology independently and facilitate the development of positron emission tomography (PET) tracers as well as therapeutic interventions for prion diseases. This is possible with cryo-EM. In a study entitled “Cryo-EM structures of prion protein filaments from Gerstmann-Sträussler-Scheinker disease”, investigators studied brain fibrils isolated from two patients with the PRNP F198S mutation (Hallinan et al., 2022). One patient was homozygous (VV) and the other homozygous (VM) at codon 129, which is known to influence disease onset and progression, allowed them to compare this important clinically relevant difference. Looking at the first VV homozygous patient, they found that the APrP was polymorphic, with mainly two, three or four intertwined protofilaments (Figure 6). The structures better delineated the location of the V residue at codon 129, which is buried deep in the amyloid core, protected from solvent exchange and tightly packed, suggesting that substitution with a methionine would affect fibril formation. These new structural data human brain APrP filaments have the potential to open new avenues for diagnosis and treatment of prion disease.
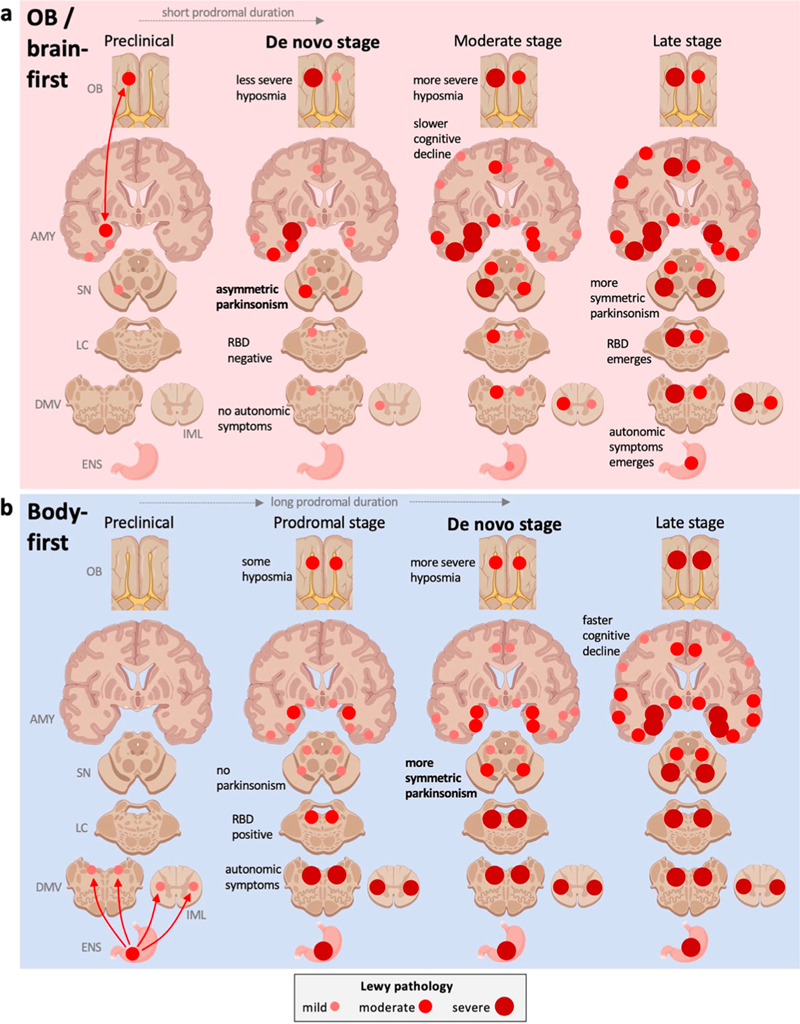
Figure 6. Analysis of the M/V 129 residue region of prion protein amyloid (APrP). a,b. Atomic models from patient 1 and 2. The 129 V residue (green) is tightly packed. c, d. This leaves little space for the larger methionine residue (reproduced with permission under the creative commons license from Hallinan et al., 2022). 7. APOE's role in chronic traumatic encephalopathy Chronic traumatic encephalopathy (CTE) results from mild yet repetitive traumatic brain injury that is often sustained during contact sports or in other contexts. While the association between this exogenous risk factor and resultant tauopathy and neurodegeneration is well established, not all participants in contact sports develop CTE. This suggests that other factors, perhaps genetic, might play a moderating role. One gene that has been implicated is APOE, which encodes the most abundant cholesterol transporter in the brain that is also the strongest genetic risk factor for sporadic AD. Previous studies have suggested that the APOEε4 allele may confer risk for a poor outcome following TBI, but the risk for CTE remains unclear. In a study by Atherton et al., investigators performed the largest study to date investigating the potential association between APOE and CTE (Atherton et al., 2022). The study was a cross sectional association analysis of 364 brain donors with repetitive head impacts exposure (i.e., contact sports or military service), with 294 positive and 70 negative for CTE. In addition to disease status, the investigators were able to explore a series of diseases relevant endophenotypes, including quantitative and semiquantitative tau pathology burden assessments. They found an association between tau pathology burden across multiple cortical regions and the amygdala as well as CTE stage among brain donors over 65 years of age at death. The presence of a APOEε4 was equivalent to 7 years of playing football. This association remained even after accounting for amyloid plaque status. These findings provide the strongest evidence to date that APOEε4 is a risk factor for CTE. 8. Unraveling genetic influences on microglia in neurodegeneration Understanding the contribution of microglia to the pathogenesis of degenerative brain disorders continues to increase in relevance. Microglia mediate numerous essential functions such as inflammation, injury repair, maintenance of brain networks, and neurodevelopment. They exhibit dynamic characteristics and can have distinct phenotypes and functions depending on environmental signals, brain regions, age, and disease pathologies. Previous studies have documented changes in microglial density, morphology, and gene expression in postmortem brain tissue from individuals with neurological and psychiatric disorders, suggesting their involvement in numerous disease processes. Recent genetic evidence has also strongly implicated myeloid cells, including microglia. To better understand the potential causal role of microglia and perhaps identify therapeutic targets, it is crucial to determine the genes influenced by disease-associated genetic variants. However, this is challenging due to the complex regulatory mechanisms involved, especially when considering variants located outside protein-coding regions. Integrating genetic and transcriptomic analyses can help identify quantitative trait loci (QTLs) and elucidate gene-gene variant relationships. Although recent efforts have detected expression QTLs (eQTLs) in microglia and their overlap with AD loci, larger sample sizes are needed. In a study conducted by Lopes et al., a team of researchers performed transcriptome analysis on 255 primary human microglial isolates obtained from multiple brain regions of 100 individuals obtained fresh at autopsy (Lopes et al., 2022). They investigated factors contributing to microglial heterogeneity, such as brain region and aging. Through mapping of expression and splicing quantitative trait loci, the study revealed that numerous susceptibility loci for neurological diseases are mediated by gene expression or splicing in microglia. Notably, the researchers identified candidate causal variants within microglia-specific enhancers, including associations between microglial expression of USP6NL and AD, as well as P2RY12 and Parkinson’s disease. Overall, this study provides a comprehensive catalog of genetic effects on the microglial transcriptome and presents potential functional variants relevant to neurological and psychiatric disorders. 9. A postmortem study examines the dual-hit hypothesis of Parkinson’s disease Pathoanatomically mapping the initiation and progression of Lewy body pathology through the brain is a critical aspect of our understanding of neurodegeneration. One hypothesis, first suggested by Braak and colleagues, is that there is a dual-hit, with simultaneous involvement of the olfactory bulb and dorsal motor nucleus of the vagus that underlies hyposmia and autonomic symptoms, respectively. Early studies had mandatory inclusion of Lewy pathology in the dorsal motor nucleus as an inclusion criterion, potentially biasing against cases with Lewy bodies in the olfactory bulb alone. Nevertheless, this hypothesis is debated and yet has spawned many clinical studies aimed at understanding potential initiating factors. Little work has been done to test the hypothesis in the neuropathological setting. In a study by Borghammer et al., investigators re-analyzed two large human post-mortem autopsy datasets that included mild Lewy body disease cases (Borghammer et al., 2022). They found that cases with alpha-synuclein immunopositivity in the peripheral autonomic nervous system and lower brainstem infrequently displayed pathology in the olfactory bulb (Figure 7). Conversely, cases with mild amygdala-predominant Lewy body pathology essentially always showed olfactory bulb pathology. Together, these findings suggest that the initiation of Lewy body pathology starts either in the olfactory bulb or enteric nervous system but rarely simultaneously. There are a number of important caveats: These cases with mild Lewy pathology were in a prodromal phase but might alternatively have not progressed. Nevertheless, these findings are an important contribution to our understanding of prion-like spreading of Lewy neuropathology and may lead to better diagnostics and advance our mechanistic understanding.
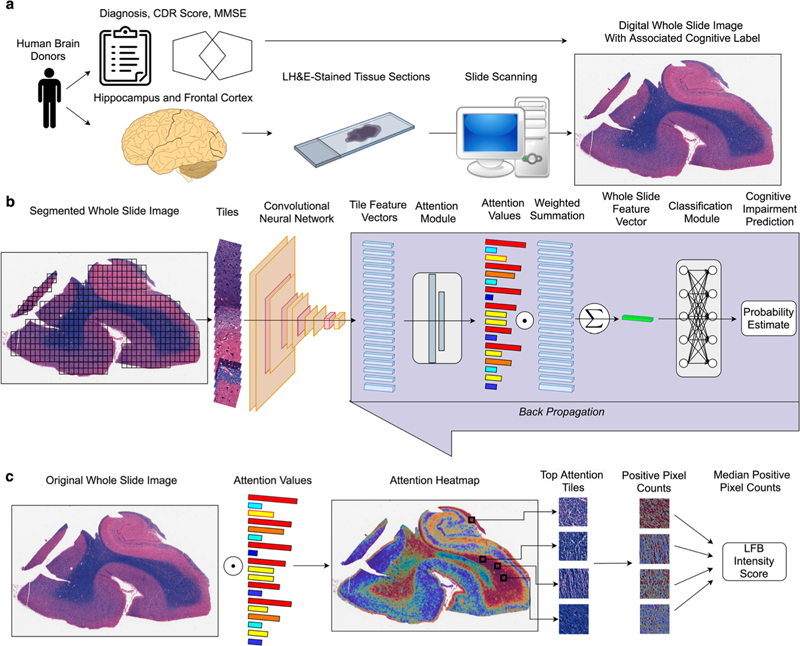
Figure 7. Single-hit hypothesis of “prion-like” propagation in Lewy body disease. a. In the “brain-first/olfactory bulb-first” subset, pathology arises in the olfactory bulb or amygdala and rapidly spreads to forebrain structures ipsilaterally and later bilaterally. b. In the “body-first” subset, pathology spreads to the medulla bilaterally and progresses superiorly. (Reproduced with permission under the creative common license from Broghammer et al., 2022). 10. Deep learning continues to reshape histological assessments Numerous studies continue to apply deep learning to digital whole slide images of histological sections of human brain tissue. This approach has been fueled by the more widespread availability of whole slide image scanners. Combined with both next generation deep learning image analysis models and powerful graphical processing units, there is fertile ground for innovation. A convolutional neural network (CNN) is a variety of model that is fine tuned for processing image data. CNNs have been applied to the detection of amyloid plaques, neurofibrillary tangles, and other features for example. These models work in a supervised fashion, requiring large numbers of annotated images for training. This is highly burdensome and acquiring this ground truth for model training represents a major barrier. In a study by McKenzie et al. (led by the author of this review), investigators applied a multiple instance learning model to a collection of whole slide images from aged individuals in a manuscript entitled “Interpretable deep learning of myelin histopathology in age-related cognitive impairment” (McKenzie et al., 2022). This model uses slide level labels, rather than annotations on patches or pixel level segmentation of individual cells (Figure 8). The investigators found that the AI was capable of predicting cognitive impairment based on a Luxol-fast blue counterstained hematoxylin & eosin-stained section alone with an accuracy of 0.65. It should be noted that this is better than chance and this is a task not currently possible by a human observer. Further analysis of the AI model found highly informative attention tiles, image patches of the whole slide image that contain the most salient aspects of the slide, were concentrated within white matter regions, not gray matter that contains well recognized structures including amyloid plaques and tangles that contribute to age-related impairment. This study paves the way towards more advanced models for analyzing relevant histological features of neurodegeneration in human autopsy brain tissues.
Figure 8. Multiple instance learning pipeline for whole slide image analysis. a. The whole slide image dataset was derived from aged human brain donors. b. Whole slide images are segmented into tiles and processed through a multiple instance learning pipeline. c. attention values are used to map salient features. (Reproduced with permission under the creative commons license from McKenzie et al., 2022). Acknowledgements This work was supported by funding from the NIH (R01AG054008, R01NS095252, R01AG062348, R01NS086736, U54NS115266, U54NS115322) as well as the Rainwater Charitable Trust/Tau Consortium, a Alexander Saint Amand Scholar Award, and a Karen Strauss Cook Research Scholar Award. Images have been reproduced under the creative commons license: visit http://creativecommons.org/licenses/by/4.0/. References 1. Atherton, K., Han, X., Chung, J., Cherry, J. D., Baucom, Z., Saltiel, N., Nair, E., Abdolmohammadi, B., Uretsky, M., Khan, M. M., Shea, C., Durape, S., Martin, B. M., Palmisano, J. N., Farrell, K., Nowinski, C. J., Alvarez, V. E., Dwyer, B., Daneshvar, D. H., … Mez, J. (2022). Association of APOE Genotypes and Chronic Traumatic Encephalopathy. JAMA Neurology, 79(8), 787–796. https://doi.org/10.1001/jamaneurol.2022.1634 2. Borghammer, P., Just, M. K., Horsager, J., Skjærbæk, C., Raunio, A., Kok, E. H., Savola, S., Murayama, S., Saito, Y., Myllykangas, L., & Van Den Berge, N. (2022). A postmortem study suggests a revision of the dual-hit hypothesis of Parkinson’s disease. NPJ Parkinson’s Disease, 8(1), 166. https://doi.org/10.1038/s41531-022-00436-2 3. Chang, A., Xiang, X., Wang, J., Lee, C., Arakhamia, T., Simjanoska, M., Wang, C., Carlomagno, Y., Zhang, G., Dhingra, S., Thierry, M., Perneel, J., Heeman, B., Forgrave, L. M., DeTure, M., DeMarco, M. L., Cook, C. N., Rademakers, R., Dickson, D. W., … Fitzpatrick, A. W. P. (2022). Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell, 185(8), 1346-1355.e15. https://doi.org/10.1016/j.cell.2022.02.026 4. Hallinan, G. I., Ozcan, K. A., Hoq, M. R., Cracco, L., Vago, F. S., Bharath, S. R., Li, D., Jacobsen, M., Doud, E. H., Mosley, A. L., Fernandez, A., Garringer, H. J., Jiang, W., Ghetti, B., & Vidal, R. (2022). Cryo-EM structures of prion protein filaments from Gerstmann-Sträussler-Scheinker disease. Acta Neuropathologica, 144(3), 509–520. https://doi.org/10.1007/s00401-022-02461-0 5. Jiang, Y. X., Cao, Q., Sawaya, M. R., Abskharon, R., Ge, P., DeTure, M., Dickson, D. W., Fu, J. Y., Ogorzalek Loo, R. R., Loo, J. A., & Eisenberg, D. S. (2022). Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature, 605(7909), 304–309. https://doi.org/10.1038/s41586-022-04670-9 6. Johnson, E. C. B., Carter, E. K., Dammer, E. B., Duong, D. M., Gerasimov, E. S., Liu, Y., Liu, J., Betarbet, R., Ping, L., Yin, L., Serrano, G. E., Beach, T. G., Peng, J., De Jager, P. L., Haroutunian, V., Zhang, B., Gaiteri, C., Bennett, D. A., Gearing, M., … Seyfried, N. T. (2022). Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nature Neuroscience, 25(2), 213–225. https://doi.org/10.1038/s41593-021-00999-y 7. Lopes, K. de P., Snijders, G. J. L., Humphrey, J., Allan, A., Sneeboer, M. A. M., Navarro, E., Schilder, B. M., Vialle, R. A., Parks, M., Missall, R., van Zuiden, W., Gigase, F. A. J., Kübler, R., van Berlekom, A. B., Hicks, E. M., Bӧttcher, C., Priller, J., Kahn, R. S., de Witte, L. D., & Raj, T. (2022). Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nature Genetics, 54(1), 4–17. https://doi.org/10.1038/s41588-021-00976-y 8. Ma, X. R., Prudencio, M., Koike, Y., Vatsavayai, S. C., Kim, G., Harbinski, F., Briner, A., Rodriguez, C. M., Guo, C., Akiyama, T., Schmidt, H. B., Cummings, B. B., Wyatt, D. W., Kurylo, K., Miller, G., Mekhoubad, S., Sallee, N., Mekonnen, G., Ganser, L., … Gitler, A. D. (2022). TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature, 603(7899), 124–130. https://doi.org/10.1038/s41586-022-04424-7 9. McKenzie, A. T., Marx, G. A., Koenigsberg, D., Sawyer, M., Iida, M. A., Walker, J. M., Richardson, T. E., Campanella, G., Attems, J., McKee, A. C., Stein, T. D., Fuchs, T. J., White, C. L., PART working group, Farrell, K., & Crary, J. F. (2022). Interpretable deep learning of myelin histopathology in age-related cognitive impairment. Acta Neuropathologica Communications, 10(1), 131. https://doi.org/10.1186/s40478-022-01425-5 10. Nelson, P. T., Brayne, C., Flanagan, M. E., Abner, E. L., Agrawal, S., Attems, J., Castellani, R. J., Corrada, M. M., Cykowski, M. D., Di, J., Dickson, D. W., Dugger, B. N., Ervin, J. F., Fleming, J., Graff-Radford, J., Grinberg, L. T., Hokkanen, S. R. K., Hunter, S., Kapasi, A., … Schneider, J. A. (2022). Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: Combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathologica, 144(1), 27–44. https://doi.org/10.1007/s00401-022-02444-1 11. Roemer, S. F., Grinberg, L. T., Crary, J. F., Seeley, W. W., McKee, A. C., Kovacs, G. G., Beach, T. G., Duyckaerts, C., Ferrer, I. A., Gelpi, E., Lee, E. B., Revesz, T., White, C. L., Yoshida, M., Pereira, F. L., Whitney, K., Ghayal, N. B., & Dickson, D. W. (2022). Rainwater Charitable Foundation criteria for the neuropathologic diagnosis progressive supranuclear palsy. Acta Neuropathologica,144(4), 603-614. https://doi.org/10.1007/s00401-022-02479-4 12. Schweighauser, M., Arseni, D., Bacioglu, M., Huang, M., Lövestam, S., Shi, Y., Yang, Y., Zhang, W., Kotecha, A., Garringer, H. J., Vidal, R., Hallinan, G. I., Newell, K. L., Tarutani, A., Murayama, S., Miyazaki, M., Saito, Y., Yoshida, M., Hasegawa, K., … Scheres, S. H. W. (2022). Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature, 605(7909), 310–314. https://doi.org/10.1038/s41586-022-04650-z
Copyright: © 2023 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |