|
Contents
1. New Diagnostic Methods
1.01 Methylthioadenosine phosphorylase immunostaining as a surrogate marker for CDKN2A/B homozygous deletion in gliomas
1.02 Artificial intelligence in morphomolecular analysis of glioblastoma
1.03 Histological and molecular correlates of TSPO labelling in human brain tissue
1.04 Towards swift and accessible precision CNS tumour diagnostics using third generation sequencing and deep transfer learning
1.05 Retinal pathology as potential biomarker of symptom severity and impairment in patients with stiff person syndrome
1.06 Deep learning based cerebrospinal fluid diagnostics
1.07 Proteomic profiling of IDH-mutant gliomas identifies HIP1R/Vimentin as surrogate markers for 1p/19q codeletion and enables prediction of chromosomal copy number variations
1.08 Cellular digital neuropathology
2. Neurooncology
2.01 Using Spatial Transciptomics for Diagnostic Analysis of Glioma
2.02 Single cell DNA amplicon sequencing reveals order of mutational acquisition in TRAF7 and KLF4 or AKT1 co-mutated meningiomas
2.03 Alterations in PTPN11 and other Noonan syndrome associated MAP-kinase signaling pathway genes accumulate in histopathologically atypical Ganglioglioma with adverse postsurgical outcome
2.04 Molecular refinement of pilocytic astrocytoma in adult patients
2.05 Exploration of cellular origins and therapeutic targets by modeling high grade pediatric glioma of the MYCN subclass in mice
2.06 The genomic and transcriptional landscape of primary central nervous system lymphoma
2.07 PMolecular mechanisms of therapy resistance in malignant melanoma brain metastasis
2.08 A peripheral nerve sheath tumor syndrome caused by postzygotic ERBB2 mutations
2.09 CNS-tumor patients within the IMPRESS-Norway trial: First year experiences
3. Neurodegeneration
3.01 CNN-supported quantification of fat compartments at abdominal MRI applied to ALS patients
3.02 Neurodegenerative iron storage disease (neuroferritinopathy) caused by a novel frameshift mutation in the ferritin heavy chain gene (FTH1 c.341-342del)
3.03 The contribution of LATE-NC to neuron loss, granulovacuolar degeneration and dementia in Alzheimer’s disease
3.04 The role of C3 inhibition in an iPSC NMJ model of neuroinflammation
3.05 Fast-track procedure for the neuropathological assessment of neurodegenerative diseases
3.06 Neurodegeneration in HSAN1 due to ATL1 (Gly66Gln) mutation is associated with defective ER- protein quality control and compromised autophagy
3.07 Single-Nucleus Chromatin Accessibility Profiling in Four-repeat Tauopathies
3.08 Application of a human stem cell transplantation model of Alzheimer’s disease to examine disease-associated changes at a single cell level in vivo
4. Neuroinflammation
4.01 Pathological And Genetic Characterization Of JC Virus Encephalopathy With An Eleven-Year-Long Disease Course
4.02 Reduction of oligodendrocyte populations in patients with late-onset multiple sclerosis
4.03 Schwann cell remyelination is a salient feature of spinal NMO with neuroprotective potential
5. Muscle / Nerve
5.01 Molecular profiling of skeletal muscle in infantile, juvenile and adult patients with Pompe disease
5.02 Expression of immune regulating proteins in skeletal muscle of different idiopathic inflammatory myopathies (IIM) subtypes
5.03 Long Term Safety and Efficacy Outcomes for X-Linked Myotubular Myopathy (XLMTM) with Gene Replacement Therapy, Resamirigene Bilparvovec (ASPIRO): Preliminary Results from Cohort 1 in ASPIRO, a Phase 1/2/3 Study
5.04 Lymphotoxin-driven chronic mucle inflammation interdepends with impaired autophagy, self-perpetuates and models inclusion body myositis in mice
5.05 Novel form of congenital myopathy caused by bi-allelic mutations in uncoordinated mutant number-45 myosin chaperone B
6. Free Topics
6.01 Deep genotype-phenotype analysis of Focal Cortical Dysplasia type 2 differentiates between a GATOR-positive autophagy altered subtype 2a and MTOR-positive migration deficit subtype 2b
6.02 Age-dependent increase of perineuronal nets in the human hippocampus of patients with and without temporal lobe epilepsy
6.03 Vakuolisierung der Dura als nicht-lymphassoziierte Veränderung
6.04 MOGHE with or without SLC35A2 brain somatic mutations reveal a common phenotype of oligodendroglial regeneration and remyelination
1. New Diagnostic Methods
1.01
Free Neuropathol 3:20:5
Methylthioadenosine phosphorylase immunostaining as a surrogate marker for CDKN2A/B homozygous deletion in gliomas
Theoni Maragkou1, Ekkehard Hewer1,2, Erik Vassella1, Baptiste Pasquier1, Stefan Reinhard1, Maja Neuenschwander1, Philippe Schucht3
1 University of Bern, Institute of Pathology, Institute of Pathology, Bern, Switzerland
2 Lausanne University Hospital, Institute of Pathology, Institute of Pathology, Lausanne, Switzerland
3 Inselspital, Bern University Hospital, Dept. of Neurosurgery, Dept. of Neurosurgery, Bern, Switzerland
Background: Homozygous deletion (HD) of the CDKN2A/B locus has emerged as an unfavorable prognostic marker in diffuse gliomas, both IDH-mutant and IDH-wildtype. Testing for CDKN2A/B deletions can be performed by a variety of approaches, including copy number variation (CNV) analysis based on genome-wide DNA methylation data, next generation sequencing (NGS) or fluorescence in-situ hybridization (FISH), but questions remain regarding the accuracy of and correlation between different testing modalities.
Aims: In this study, we assessed the utility of S-methyl-5'-thioadenosine phosphorylase (MTAP) and cellular tumor suppressor protein pl61NK4a (p16) immunostaining as surrogate markers for CDKN2A/B HD in gliomas, across different histological tumor grades and IDH mutation status.
Question: Are MTAP and p16 accurate surrogate markers for CDKN2A/B HD in gliomas?
Methods: IDH1 R132H, ATRX and MTAP immunohistochemistry was performed on tissue microarrays (TMAs) of 301 diffuse gliomas. Survival analysis was performed to assess the prognostic value of MTAP. Furthermore, 100 consecutive cases of gliomas were collected, in order to correlate MTAP and p16 expression with the CDKN2A/B status in CNV plot of each tumor.
Results: MTAP deficiency was associated with shortened survival in IDH-mutant astrocytomas (n=75; median survival 61 vs. 137 months; p<0.0001), IDH-mutant oligodendrogliomas (n=59; median survival 41 vs. 147 months; p<0.0001) and IDH-wildtype gliomas (n=117; median survival 13 vs. 16 months; p=0.011). In a cohort of 100 gliomas, complete loss of MTAP and p16 by immunohistochemistry was 100 % and 90 % sensitive as well as 97 % and 89 % specific for CDKN2A/B HD, respectively, as identified on CNV plot derived from genome-wide DNA methylation analysis. Two cases with MTAP and p16 loss of expression did not demonstrate CDKN2A/B HD in CNV plot, however FISH analysis confirmed the HD for CDKN2A/B.
Conclusions: MTAP immunostaining is an important complement for diagnostic work-up of gliomas, because of its excellent correlation with CDKN2A/B status, robustness, rapid turnaround time and low-costs, while p16 immunostaining represents a good alternative for detecting CDKN2A/B HD. Discovering CDKN2A/B HD through MTAP and/or p16 immunohistochemistry seems to be a more accurate method than the CNV analysis derived from genome-wide DNA methylation data.
1.02
Free Neuropathol 3:20:6
Artificial intelligence in morphomolecular analysis of glioblastoma
Stephan Balogh1, Karen Brengmann1, Jannik Sehring1, Thomas Kauer1, Gudrun Schmidt1, Till Acker1, Daniel Amsel1, Hildegard Dohmen1
1 Institute of Neuropathology, Justus Liebig University Giessen, Giessen, Deutschland
Background: Glioblastoma is the most common primary malignant brain tumor and has a poor prognosis despite existing treatment options. It is characterized by its inhomogeneous appearance and molecular heterogeneity. A detailed diagnosis is desirable, especially with regard to the emerging personalized medicine to guide treatment decisions. The increasing application of whole slide image scanners enables the digitalization of histopathological slides, collected in clinical routine diagnostics, into high-quality images that offer new possibilities for computer-aided precision diagnostics.
Objective: The goal of our work-in-progress project is to curate a high-resolution dataset with annotations to train an artificial intelligence to independently recognize characteristic structures of glioblastoma tissue sections, such as tumor area, vascular proliferation and necrosis. Using these AI-based algorithms, we aim to gain new insights that could help refine the characterization of glioblastomas by correlating morphological information with available clinical and molecular data.
Methods: Initially, clinical and molecular information on 200 glioblastoma patients was gathered. The corresponding hematoxylin-eosin-stained histopathological slides were then digitized using a high-throughput whole slide image scanner (Hamamatsu NanoZoomer S360). The annotation of key features in the images was divided into two distinct phases. Prior to the actual annotation phase, two MD students completed a training phase with a small batch of images (n=10) in order to get hands-on experience with the annotation software and difficult issues (QuPath version 0.3.0). The ground truth was determined by an experienced neuropathologist.
Evaluation: The inter- and intraobserver variability of the two students will be evaluated with regard to the learning progress (e.g. accuracy, classification of a tissue area) and differences between the annotations of the two students, derived from subjective assessment. In the second part, correlations between morphological information (vascular proliferation and necrosis with pseudopalisading) and clinical and molecular information will be examined. These annotated slides will serve as training and test sets for in-house AI-based predictions.
Perspective: Our data will be used as a resource for an in-house developed app that will serve as a learning solution for medical students, but also as a crowdsourcing platform for the annotation of features on small patches of whole slide images. In addition, the curated high-resolution data set will serve as input for further internally developed AI algorithms to support tumor diagnosis and therapy decisions.
1.03
Free Neuropathol 3:20:7
Histological and molecular correlates of TSPO labelling in human brain tissue
Lorraine Weidner1,2, Franziska Dekorsy3, Stefanie Quach4, Viktoria Ruf5, Julia Lorenz1,2, Peter Hau2,6, Jörg-Christian Tonn4,5, Peter Bartenstein3,7, Matthias Brendel3,7, Nathalie L. Albert3,7, Markus J. Riemenschneider1,2
1 Regensburg University Hospital, Department of Neuropathology, Regensburg, Deutschland
2 Regensburg University Hospital, Wilhelm Sander Neuro-Oncology Unit, Regensburg, Deutschland
3 University Hospital of Munich, LMU Munich, Department of Nuclear Medicine, München, Deutschland
4 University Hospital of Munich, LMU Munich, Department of Neurosurgery, München, Deutschland
5 LMU Munich, Center for Neuropathology and Prion Research, München, Deutschland
6 Regensburg University Hospital, Department of Neurology, Regensburg, Deutschland
7 German Cancer Research Center (DKFZ), German Cancer Consortium (DKTK), partner site Munich, Heidelberg, Deutschland
Background: TSPO is frequently upregulated in neoplastically transformed tissues, including glioblastomas. This may be of use for PET imaging of brain tumors. However, due to the heterogeneity of cell populations that could contribute as TSPO-PET signal source in gliomas, the imaging biomarker interpretation may be challenging.
Aim: We therefore dissect TSPO labelling in connection with the underlying histopathological and molecular features in biopsy samples from glioma patients.
Question: To decipher the underlying histopathological and molecular features of TSPO-PET enrichment.
Methods: We aim to collect a total of 75 glioma patients all characterized by MRI, TSPO- and FET-PET. TSPO protein expression and expression of cell differentiation markers are assessed immunohistochemically on consecutive sections and by multiplex stains. RNA isolation has been optimized to perform RNA-Seq on biopsy samples and to compare regions of high and low TSPO-PET signal/protein expression. To identify relevant hallmarks and GO terms we use DESeq2 followed by FUMA and Reactome as well as GSEA with normalized counts. Furthermore, exceeding the biopsy study we stain tissue microarrays for TSPO that cover a broader spectrum of human brain pathologies as well as a spectrum of non-neoplastic tissues from different brain regions. To better understand TSPO regulation, we consult data of large patient cohorts from the TCGA database, perform in vitro epigenetic investigations on AZA- or TSA-treated patient-derived glioblastoma cell lines and analyze the TSPO promoter in gliomas by direct bisulfite sequencing.
Results: We report the interim analysis of the glioma patients that have been included and fully histologically characterized in the biopsy study so far. Our results suggest that (apart from microglia and macrophages) the glial tumor cells relevantly contribute to the overall TSPO signal in these patients. RNA-Seq analyses comparing TSPO high and low regions (both by PET and protein expression) indicate three TSPO-dependent functional clusters, i.e apoptosis/DNA repair, extracellular matrix organization and immune system. Furthermore the tissue microrarrays show heterogeneity of TSPO expression between different brain pathologies and non-neoplastic brain regions. Bringing this information together with TSPO-PETs from respective patients/brain regions will generate a map of TSPO expression in healthy and diseased brain for clinical use. Finally, our epigenetic investigations suggest that a loss of TSPO methylation in high-grade neoplasms may mechanistically contribute to the TSPO overexpression observed in these tumors.
Conclusion: Taken together, our approach of integrating histological, molecular and imaging data will provide unique insights into TSPO-PET enrichment patterns and will help to better understand and to comprehensively describe the clinical relevance of this novel imaging biomarker.
1.04
Free Neuropathol 3:20:9
Towards swift and accessible precision CNS tumour diagnostics using third generation sequencing and deep transfer learning
Areeba Patel1,2, Helin Dogan1,2, Alexander Wolfgang Jung3, Zaira Seferbekova4, Alexander Payne5, Natalie Schoebe1,2, Elena Krause1,2, Michael Ritter1,2, Daniel Schrimpf1,2, Damian Stichel1,2, Stefan Hammelmann1,2, Christina Blume1,2, Philipp Euskirchen6, Violaine Goidts7, Martin Sill8,9, Stefan Pfister8,9, Matthew Loose5, Wolfgang Wick10,11, Andreas von Deimling1,2, David Jones8,12, Matthias Schlesner13, Moritz Gerstung3,4, Felix Sahm1,2
1 German Cancer Research Center, Clinical Cooperation Unit Neuropathology, Heidelberg, Deutschland
2 University Hospital Heidelberg, Neuropathology, Heidelberg, Deutschland
3 European Molecular Biology Laboratory, European Bioinformatics Institute EMBL-EBI, Hinxton, United Kingdom
4 German Cancer Research Center, Division of Artificial Intelligence in Oncology, Heidelberg, United Kingdom
5 University of Nottingham, DeepSeq, School of Life Sciences, Nottingham, United Kingdom
6 Charité-Universitätsmedizin Berlin, Neurology, Berlin, Deutschland
7 German Cancer Research Center, Brain Tumor Translational Targets, Heidelberg, Deutschland
8 Hopp Children's Cancer Center (KiTZ), Heidelberg, Deutschland
9 German Cancer Research Center, Division of Pediatric Neurooncology, Heidelberg, Deutschland
10 German Cancer Research Center, Clinical Cooperation Unit Neurooncology, Heidelberg, Deutschland
11 National Center for Tumor Diseases, Department of Neurology and Neurooncology Program, Heidelberg, Deutschland
12 German Cancer Research Center, Pediatric Glioma Research Group, Heidelberg, Deutschland
13 Augsburg University, Biomedical Informatics, Data Mining and Data Analytics, Augsburg, Deutschland
Background: Molecular markers are now unequivocally a requirement for integrative brain tumour diagnostics. The 2021 WHO classification of central nervous system (CNS) tumours substantially increases the set of genes required in routine evaluation, and significantly increases the relevance of DNA methylation analysis in the diagnostic process. Owing to extensive setup costs and batch requirements, smaller labs and clinics might not be able to deliver molecular results for prompt clinical decisions. Deep neural network architectures have been shown to predict whole genome duplications, driver gene mutations, transcriptomic associations, immune cell localisation and prognostic effects from H&E slides. Third generation sequencing has enabled sequencing longer reads, shorter library preparation protocols, ability to call base modifications natively, real time analysis, and low-cost, portable devices.
Aims: To make precision diagnostics accessible, we introduce an integrated computational histopathology and third generation sequencing workflow for real-time CNS tumour molecular diagnostics.
Methods: We present CNS-CHiP- a multi-task lightweight deep transfer learning model to predict key molecular alterations, methylation classification and survival from H&E stained CNS tumour slides. The model provided basic information regarding the tumor type instantly. For further detail (e.g. variant of IDH alteration) and subtyping, we subsequently used the predictions to formulate a custom panel for each patient. Targeted sequencing and analyses were performed using Rapid-CNS2- a custom neurooncology third generation sequencing pipeline for parallel copy-number profiling, mutational and methylation analysis that is highly flexible in target selection, requires no additional library preparation for targeting, runs efficiently on single samples, and can be initiated upon receipt of frozen sections. Rapid-CNS2 leverages adaptive sampling through ReadFish and was run using a portable MinION or GridION device.
Results: We show that CNS-CHiP can predict a multitude of key pathognomonic alterations (eg. IDH mutation, 7 gain/10 loss, etc.) using a single model with reasonable accuracy. Using a personalised panel for targeted sequencing of each sample enabled smaller target sizes, thus reducing sequencing time to an average of 24 hours. CNS-CHiP predictions were compared to their respective Rapid-CNS2 results and corresponding conventional data (NGS panel sequencing and EPIC array analysis). We demonstrate our workflow on prospective diagnostic samples received by the Department of Neuropathology, University Hospital Heidelberg. The average turnaround time per sample was 48h.
Conclusions: Our workflow harnessing histology-based molecular predictions to instruct targeted sequencing can be set up with low initial investment, reduces hands-on time and has the potential to facilitate reporting of integrated molecular diagnostic results in less than 48h. CNS-CHiP combined with Rapid-CNS2 thus aims to make CNS molecular diagnostics affordable and accessible to smaller hospitals and labs especially in low- and middle-income countries.
1.05
Free Neuropathol 3:20:11
Retinal pathology as potential biomarker of symptom severity and impairment in patients with stiff person syndrome
Sabine Seefried1, Claudia Sommer1
1 UKW, Würzburg, Deutschland
Background: Stiff-person syndrome (SPS) is a rare chronic autoimmune disease characterized by painful spasms and rigidity, predominantly of the axial and lower extremity muscles. Autoantibodies have been reported in 80% of cases, disrupting the function of the inhibitory neurotransmitters GABA and glycine, and leading to overexcitability of the neuromuscular system and psychological comorbidities. Since the retina is highly enriched with GABAergic neurons, retinal pathology may occur in SPS, especially in the ganglion cell and inner plexiform layer (GCIPL) of the retina, which contains most of the retinal GABAergic neurons.
Study aims: We aimed to detect potential abnormalities in the retina in SPS patients and correlate these with symptom severity and impairment.
Hypotheses: We hypothesized a lower retinal thickness in SPS patients in comparison to normal healthy controls and patients with diabetes and furthermore correlations between GAD antibody levels and findings in retina layer thickness and correlations with severity of the disease.
Methods: 24 GAD positive SPS patients (17 female; 7 male; aged 53 ±7; 9 with diabetes) received a clinical examination and optical coherence tomography (OCT) for retina layer thickness. Blood was drawn for autoantibody detection. Data were compared to a matched healthy cohort and a matched patients group with diabetes but no diabetic polyneuropathy. The severity of SPS symptoms and impairment was assessed on the basis of the ability to walk: free walking, use of a crutch, use of two crutches, walking on the rollator or sitting in a wheelchair.
Results: OCT showed lower retinal thickness in GCIPL and lower average macular thickness (AMT) in the SPS patients (GCIPL: 73.34±5,3; AMT: 302.98 ±12,1) in comparison to healthy controls (GCIPL: 76.01 ± 4,2; AMT: 311.76±13,4). Comparison of the 15 SPS patients without diabetes with the total number of 25 SPS patients showed no difference between these results. The control patients with only diabetes had no abnormalities in the OCT results compared to the healthy controls, indicating that the atrophy of GCIPL and AML is likely to be caused exclusively by the SPS and not by diabetes. There was a positive correlation between walking ability and GCIPL thickness in SPS patients; higher GCIPL thickness was associated with better walking ability (R² = 0,92).
Conclusion: This study indicates retinal involvement in SPS. OCT might be useful as a complementary diagnostic tool in SPS, and retinal layer thickness measurements might be developed as a non-invasive biomarker for disease progression.
1.06
Free Neuropathol 3:20:12
Deep learning based cerebrospinal fluid diagnostics
Leonille Schweizer1,2, Philipp Seegerer3, Hee-Yeong Kim4, René Saitenmacher3, Amos Münch1, Liane Barnick1, Anja Osterloh1, Carsten Dittmayer1, Ruben Jödicke1, Debora Pehl1, Annekathrin Reinhardt5, Klemens Ruprecht6, Annika K Wefers7, Patrick Harter8, Ulrich Schüller7, Frank L Heppner1, Maximilian Alber3, Klaus-Robert Müller3, Frederick Klauschen9
1 Institute of Neuropathology, Charité – Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin and Berlin Institute of Health, Berlin, Berlin, Deutschland
2 German Cancer Consortium (DKTK), Partner Site Berlin, and German Cancer Research Center (DKFZ), Berlin, Deutschland
3 Machine-Learning Group, Department of Software Engineering and Theoretical Computer Science, Technical University of Berlin, Berlin, Deutschland
4 Systems Medicine of Infectious Disease, Robert Koch Institute, Berlin, Deutschland
5 Department of Neuropathology, University Hospital Heidelberg, Heidelberg, Deutschland
6 Department of Neurology, Charité University Medicine Berlin, Berlin, Deutschland
7 Institute of Neuropathology, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
8 Neurological Institute (Edinger Institute), Goethe University, Frankfurt, Deutschland
9 Institute of Pathology, Ludwig-Maximilians-Universität München, München, Deutschland
Background: The analysis of cerebrospinal fluid (CSF) specimens is essential for the diagnostic workup and clinical management of neurological patients and relies on differential cell typing. Because blood cell cytometers are unable to identify diagnostically relevant cell types in CSF samples, the current gold-standard is based on microscopic examination by specialized technicians and neuropathologists. Manual differential cell count is time-consuming, labor-intensive and subjective. We therefore set out to compile a real-world CSF dataset including all diagnostically relevant cell types to train a robust algorithm for cell type differentiation with the potential to solve complex diagnostic tasks.
Methods: We therefore developed an image analysis approach based on expert annotations of 127.455 digitized CSF objects from 78 patients corresponding to 15 clinically relevant categories and trained a multiclass convolutional neural network (CNN). We applied explainable artificial intelligence (xAI) methods to elucidate the most relevant image pixels for CNN predictions and compare pattern recognition to humans. We further developed a new data partitioning strategy for further machine learning projects. To assess the realistic usefulness in diagnostic practice, we validated the CNN-based approach by comparing its performance to that of seven board certified neuropathologist from different academic institutions.
Results: The CNN classified the 15 categories with high accuracy (mean AUC 97.3%). By using xAI, we could demonstrate that the CNN identified meaningful substructures in CSF cells recapitulating human pattern recognition. We validated the diagnostic performance of the CNN by comparing the predictions of 511 cells selected from 12 different CSF samples to seven board-certified neuropathologists blinded for clinical information. Inter-rater agreement between the CNN and the ground truth was non-inferior (Krippendorff’s alpha 0.79) compared to the agreement of seven human raters and the ground truth (mean Krippendorff’s alpha 0.72, range 0.56-0.81). The CNN assigned the correct diagnostic label (inflammatory, hemorrhagic or neoplastic) in 10 out of 11 clinical samples compared to 7-11 out of 11 correctly labeled CSFs by human raters. Similar to four human raters, the CNN misclassified single highly activated B-cells as cancer cells in two samples, but indicated reduced confidence by low predicted probability vectors for the difficult cases.
Conclusions: Our approach not only provides the basis to overcome current limitations in automated cell classification for routine diagnostics, but also demonstrates how a visual explanation framework can connect machine decision-making with cell properties and thus provide a novel versatile and quantitative method for investigating CSF manifestations of various neurological diseases.
1.07
Free Neuropathol 3:20:14
Proteomic profiling of IDH-mutant gliomas identifies HIP1R/Vimentin as surrogate markers for 1p/19q codeletion and enables prediction of chromosomal copy number variations
Marius Felix1, Dennis Friedel1, Ashok Kumar Jayavelu2, Katharina Filipski3, Anne-Kathrin Reinhard1, Uwe Warnken4, Damian Stichel1, Daniel Schrimpf1, Andrey Korshunov1, Yueting Wang1, Tobias Kessler4, Nima Etminan5, Andreas Unterberg6, Christel Herolod-Mende6, Laura Heikaus7, Felix Sahm1, Wolfgang Wick4, Patrick N. Harter3, Andreas von Deimling1, David E. Reuss1
1 Department of Neuropathology, Institute of Pathology, Heidelberg, Deutschland
2 Clinical Cooperation Unit Pediatric Leukemia, German Cancer Research Center (DKFZ), Heidelberg, Deutschland
3 Institute of Neurology, Edinger Institute, Heidelberg, Deutschland
4 Clinical Cooperation Unit Neurooncology, German Consortium for Translational Cancer Research (DKTK), German Cancer Research Center (DKFZ), Heidelberg, Deutschland
5 Department of Neurosurgery, Mannheim, Deutschland
6 Department of Neurosurgery, Heidelberg, Deutschland
7 Bruker GmbH, Bremen, Deutschland
Background: IDH-mutant gliomas are a common but heterogenous group of diffuse gliomas. Chromosomal copy number variations (CNV) are a hallmark of many different types of cancer and 1p/19q codeletion is mandatory to differentiate “astrocytoma, IDH mutant” from “oligodendroglioma, IDH-mutant and 1p/19q-codeleted”. Currently, loss of nuclear ATRX is the only surrogate marker for an 1p/19q-wildtype status accepted by WHO. Unmet diagnostic needs are improved surrogate markers for 1p/19q codeletion in ATRX retaining gliomas and a rapid determination of CNVs in general.
Aims: We aimed at the identification of protein-level surrogate markers for 1p/19q codeletion suitable for immunohistochemical assays as well as proteomic signatures associated with chromosomal alterations in general.
Methods: We used mass-spectrometry (MS) based proteomics to analyze IDH-mutant gliomas pre-characterized by DNA methylation profiling. A discovery series containing 35 fresh frozen (FF) and 72 formalin fixed and paraffin embedded (FFPE) tumors were analyzed and potential biomarkers for 1p/19q codeletion were identified. A subsequent validation series consisting of 50 oligodendrogliomas and 50 astrocytomas was evaluated using immunohistochemistry to confirm potential biomarker identifications based on proteomic discoveries. Furthermore, an additional validation cohort of 69 IDH-mutant gliomas was stained and evaluated in a separate institution.
Results: Proteomic data from FF and FFPE tissues were comparable. Highly specific protein patters were identified, which could distinguish between oligodendroglioma and astrocytoma. Oligodendrogliomas showed high HIP1R and low vimentin (VIM) staining intensities and astrocytomas low HIP1R and high VIM staining intensities. Blinded evaluation of the validation cohort revealed a specificity of 100% and sensitivity 90-94% between two observers for 1p/19q prediction. Combined evaluation with ATRX increased sensitivity to 96%. An additional verification cohort stained and evaluated in a separate institution revealed similar prediction performances. Further analyses revealed that a high proportion of the differentially regulated proteins between astrocytoma and oligodendroglioma are coded on the 1p and 19q chromosome arms. By generating virtual protein abundance means from unregulated chromosome arms, chromosomal protein ratios (CPRs) were calculated, which helped predict copy number variations, showing high correlation with CNV plots from genome wide DNA methylation profiles.
Conclusions: MS based analysis of FFPE tissue highly correlates with FF tissue, allowing in depth differential proteomic profiling. Proteomics enables the discovery of new biomarkers and has great potential for the future of brain tumor diagnostics. Immunohistochemistry for HIP1R, VIM and ATRX can predict 1p/19q status with high specificity and sensitivity. CPRs are a promising tool for the rapid proteome-based determination of chromosomal copy number variations.
1.08
Free Neuropathol 3:20:16
Cellular digital neuropathology
Jonas Franz1, Christine Stadelmann1
1 Institut für Neuropathologie, Universitätsmedizin Göttingen, Göttingen, Deutschland
Background: Neuropathology is traditionally based on histological analysis of tissue. Aside molecular pathology also the introduction of digital microscopy is changing the working environment of neuropathologists. Digital pathology comes with modern algorithms for image quantification. Mostly these algorithms are capable of solving sophisticated classification or segmentation problems. While classification is often applied to whole disease entities and segmentation is used to find, e.g., immunpositive areas we tried to adopt the algorithms to the traditional concept of „Cellularpathologie“ as defined by R. Virchow.
Aims: We aimed at optimizing existing image analysis algorithms and concepts specifically to the need of classification of cells on whole slide images.
Question: The main question was to determine a workflow which integrates on the one hand the needs of neuropathologists to steer the analysis and on the other hand to implement even advanced computer technology, e.g., with deep learning-based analyses.
Method: Our main method focused on supervised deep learning to classify single cells in immunfluorescence stainings based on existing nuclear segmentation algorithms.
Result: As a result we deployed locally an image classification server for cell annotation by neuropathological experts without background in computer science in combination with an open microscopy (omero) server. Manually annotated images were used to train various classification algorithms in combination with data augmentation and other techniques to optimize performance. After model selection we could reach almost human performance (>98% accuracy with <2-3% false positive rate) in an exemplary project of microglial cell classification stained by iba1.
Conclusion: We conclude that this concept of image analysis generalizes to various sorts of immunfluorescence stainings and might thus help to elevate the single cellular analysis to a whole tissue-based analysis with millions of cells.
2. Neuroonocology
2.01
Free Neuropathol 3:20:17
Using Spatial Transciptomics for Diagnostic Analysis of Glioma
Michael Ritter1,2, Christina Blume1,2, Areeba Patel1,2, Philipp Sievers1,2, Helin Dogan1,2, Christel Herold-Mende3, Wolfgang Wick4,5,6, Andreas von Deimling1,2, Felix Sahm1,2
1 University Hospital Heidelberg, Department of Neuropathology, Heidelberg, Deutschland
2 German Cancer Research Center (DKFZ), German Consortium for Translational Cancer Research (DKTK), Clinical Cooperation Unit Neuropathology, Heidelberg, Deutschland
3 University Hospital Heidelberg, Department of Neurosurgery, Heidelberg, Deutschland
4 German Cancer Research Center (DKFZ), German Consortium for Translational Cancer Research (DKTK), Clinical Cooperation Unit Neurooncology, Heidelberg, Deutschland
5 University Hospital Heidelberg, Neurology Clinic, Heidelberg, Deutschland
6 German Cancer Research Center (DKFZ), National Center for Tumor Diseases (NCT), Department of Neurology and Neurooncology Program, Heidelberg, Deutschland
Background: The lack of specific in-situ markers of IDH-wildtype glioblastoma makes it hard to distinguish between infiltrating tumour cells and reactive tissue. Determination of copy number variations (CNV) of chromosomes may assist in this diagnostic challenge, but often requires a large amount of tissue, which is not always available. Especially for stereotactic biopsies the available material is often quite limited.
Aims: In our study we wanted to test the feasibility of using spatial transcriptomics to distinguish between invading tumour cells and reactive/adjacent tissue and if it is possible to generate useful data like CNVs from small tissue fragments.
Methods: We applied spatial transcriptomics on 12 FFPE GBM samples indicating reactive tissue or an infiltration zone and 4 stereotactic biopsies. We determined the CNV for all samples and used a single cell dataset of infiltrating tumour cells (Darmanis et al., 2017) to map the different cell types onto the tissue.
Results: The mapping of the tumour cells onto the sections clearly distinguished the tumour from the adjacent tissue and also allowed to further distinguish between majorly reactive tissue and tissue with a high number of infiltrating tumour cells. Nevertheless, mapping on single cell level resolution requires higher resolution methods. Both mapping by Chr. 7 gain and Chr. 10 loss and expression-based mapping produced similar results. Using spatial transcriptomics on stereotactic biopsies we were able to determine the major CNVs from a 5 µm thick tissue section with a 1 mm diameter.
Conclusion: In summary we prove the feasibility to identify the infiltration zone and distinguish from reactive tissue using spatial transcriptomics, and we were able to derive CNV from very small tissue fractions. This can be leveraged especially if immunohistochemical stainings are not informative or too little tissue is left for the determination of CNV profiles.
2.02
Free Neuropathol 3:20:18
Single cell DNA amplicon sequencing reveals order of mutational acquisition in TRAF7 and KLF4 or AKT1 co-mutated meningiomas
Helin Dogan1, Christina Blume1, Areeba Patel1, Gerhard Jungwirth2, Miriam Ratliff3, Ralf Ketter4, Wolfgang Wick5, Christel Herold-Mende2, David Reuss1, Andreas von Deimling1, Felix Sahm1
1 Clinical Cooperation Unit Neuropathology, German Cancer Consortium (DKTK), German Cancer Research Center and Dept. of Neuropathology, University Hospital Heidelberg, Heidelberg, Deutschland
2 Dept. of Neurosurgery, University Hospital Heidelberg, Heidelberg, Deutschland
3 Dept. of Neurosurgery, University Hospital Mannheim, Mannheim, Deutschland
4 Dept. of Neurosurgery, University Hospital Saarland, Homburg, Deutschland
5 Dept. of Neurology and Neurooncology Program, National Center for Tumor Diseases, University Hospital Heidelberg, Heidelberg, Deutschland
Background: Most meningiomas carry mutations in the tumor suppressor neurofibromatosis gene 2 (NF2) on chromosome 22q, while NF2-wildtype meningiomas account for about one third of all. In non-NF2-mutated cases, SMO, POLR2A, PIK3CA, AKT1 and KLF4 mutations, the latter both typically with TRAF7 mutations, have been described. The combination of AKT1 and KLF4, respectively, with TRAF7 is intriguing: TRAF7/AKT1 co-mutations are associated with meningothelial histology and basal localization, while TRAF7/KLF4 co-mutations are highly specific for secretory meningioma without any predominant localization. Since bulk molecular profiling indicates a step-wise mutational acquisition, the mutational sequence, whether the alteration in TRAF7 or in AKT1/KLF4 occurs first, has remained elusive.
Methods: Single-cell sequencing technologies have allowed direct insight into the clonal architecture and complexity of thousands of individual cells. After evaluation of a patient with two independent meningiomas having identical somatic TRAF7 mutation but separate AKT1/KLF4 hotspot mutation, variant allele frequencies (VAFs) of 62 retrospectively collected meningiomas carrying either co- or single-mutations in TRAF7 and/or AKT1 or KLF4 were compared using bulk hybrid-capture panel sequencing data. Additionally, a custom tumor panel comprising 392 amplicons covering 28 genes as well as the TERT promoter was used along with the amplicon-based Tapestri technology for single cell DNA sequencing. Genotype clustering analysis was finally performed to reveal the order of mutational acquisition in our cohort of TRAF7mut/AKT1mut and TRAF7mut/KLF4mut meningiomas (n=7).
Results: Looking at mutational co-occurrence in bulk data, mutations assigned with higher VAFs, unless explained by copy number changes, are thought to be acquired earlier than those with lower VAFs. Our bulk data of 28 co-mutated cases showed no significant difference in bulk-measured VAFs, suggesting there was no major gap between the two time points of mutational acquisition. However, the majority of single-mutated cases (21/36) harbored mutations in TRAF7, while the others were either only AKT1 (n=12) or KLF4 (n=3) mutant. While it remains impossible to delineate clonal architecture from bulk data, our single cell data allowed grouping of cells into clonal populations. A total of 875,000 cells from 7 samples were prepared resulting in a median throughput of 2315 cells per sample and a median sequencing coverage of 105 reads per cell per amplicon. Our data revealed three subclones in each sample: one wildtype clone (potentially stroma cells), one clone carrying a single mutation in TRAF7 (detected for 6/7 samples) and another clone harboring the co-mutations in TRAF7 and KLF4 or AKT1.
Conclusions: Our findings strengthen the hypothesis that in TRAF7mut/AKT1mut and TRAF7mut/KLF4mut meningiomas, the mutation in TRAF7, which can occur throughout the WD40 domain of the protein, is acquired in an earlier stage than the hotspot mutation in AKT1 or KLF4. This study shows, that single-cell technologies on DNA are useful in elucidating clonal architecture and phylogenetic trees. Although single-cell DNA sequencing in particular is associated with technical challenges such as false positive variant calling and allelic dropouts, high numbers of recovered cells as well as high-quality sequencing allow conclusive information on cellular zygosity and a robust analysis of mutational acquisition.
2.03
Free Neuropathol 3:20:20
Alterations in PTPN11 and other Noonan syndrome associated MAP-kinase signaling pathway genes accumulate in histopathologically atypical Ganglioglioma with adverse postsurgical outcome
Lucas Hoffmann1, Roland Coras1, Katja Kobow1, Javier Lopez-Riviera2,3,4, Costin Leu3,4,5,6, Dennis Lal3,4,5,6, Peter Nürnberg6, Christian G. Bien7, Thilo Kalbhenn7, Markus Müller7, Hajo Hamer8, Sebastian Brandner9, Karl Rössler9,10, Samir Jabari1, Ingmar Blümcke1
1 Department of Neuropathology, Universitätsklinikum Erlangen, FAU Erlangen-Nürnberg, Partner of the European Reference Network (ERN) EpiCARE , Erlangen, Deutschland
2 Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, United States
3 Genomic Medicine Institute, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, United States
4 Charles Shor Epilepsy Center, Neurological Institute, Cleveland Clinic, Cleveland, United States;
5 Stanley Center for Psychiatric Research, Broad Institute of Harvard and M.I.T, Cambridge, MA 02142, United States
6 Cologne Center for Genomics (CCG), Medical Faculty of the University of Cologne, University Hospital of Cologne, Cologne, Germany
7 Department of Epileptology (Krankenhaus Mara), Medical School, Bielefeld University, Bielefeld, Germany
8 Epilepsy Center, Universitätsklinikum Erlangen, FAU Erlangen-Nürnberg, Erlangen, Germany, and EpiCARE partner, Erlangen, Germany
9 Department of Neurosurgery, Universitätsklinikum Erlangen, FAU Erlangen-Nürnberg, Erlangen, Germany, and EpiCARE partner, Erlangen, Germany
10 Department of Neurosurgery, Medical University of Vienna, Vienna General Hospital, Vienna, Austria, Vienna, Austria
Background: The PTPN11 gene is a tyrosine phosphatase non-receptor type protein linked to the MAP kinase signaling pathway. It was recently discovered as novel lesional epilepsy gene by large exome-wide sequencing studies. PTPN11 germline mutations have been associated with Noonan syndrome, a multisystem disorder characterized by facial features, developmental delay and other organ diseases. Sporadically, low-grade epilepsy-associated brain tumors (LEAT) also occur in Noonan patients. Herein, we performed a first deep phenotype-genotype analysis of low-grade developmental brain tumours with brain somatic alterations of the PTPN11 gene as compared to commonly observed LEAT with or without MAP kinase signaling pathway alterations.
Methods: We selected 87 LEAT cases recently submitted to whole exome sequencing and genotyping including 17 dysembryoplastic neuroepithelial tumours (DNT) and 70 ganglioglioma (GG). Clinical data were retrieved from hospital files including postsurgical outcome (Engel outcome, seizure onset, age at surgery, MRI findings, location). Available histopathology slides were fully digitalized for systematic microscopy analysis, including H&E and immunohistochemistry for CD34, p16, MAP2, NeuN, Ki67, IDH1 and p53.
Results: We identified a series of eight GG with PTPN11 alterations, i.e. gains in copy number variations (CNV) of the locus 12q, which showed a systematic pattern of additional CNV gains in FGFR4, RHEB, NF1, KRAS as well as BRAF alterations (Figure 1). Histopathology pattern analysis revealed an atypical and complex glio-neuronal phenotype with subpial tumour spread and large, pleomorphic and multinuclear cellular features (Figure 2). Only three out of eight GG with PTPN11 alterations were free of disabling-seizures two years after surgery (Engel Ia outcome, 38%). This was remarkably different from our series of GG with BRAF alterations (n=35), GG without any genetic alteration detectable by our study paradigm (n=27) and DNT with FGFR1 alterations (n=6) with Engel Ia rates of 85%, 76% and 83%, respectively.
Conclusions: We identified a subgroup of ganglioglioma characterized by PTPN11 alterations in association with other Noonan syndrome related alterations of the MAP kinase signaling pathway, i.e., KRAS, RHEB, BRAF, and FGFR4. These tumours were further characterized by histopathological features of cellular atypia in glial and neuronal cell components as well as adverse postsurgical outcome. These features were strikingly different from other LEAT with defined genetic alterations in BRAF, e.g., V600E mutation, and FGFR1. Notwithstanding, these findings need further validation as they argue for a three-tiered WHO grading system also for developmental, glio-neuronal tumors associated with early-onset focal epilepsy. Genetic similarities to Noonan syndrome and Noonan syndrome associated disorders may also suggest the use of targeted treatment options against the MAP kinase and mTOR signaling pathway.
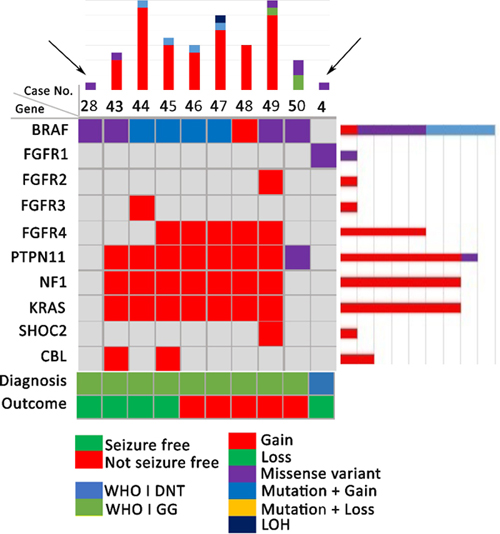
Figure 1: Oncoplot of PTPN11 altered LEAT compared to a BRAF-V600E mutated GG (arrow on left) and a FGFR1 altered DNT (arrow on right)
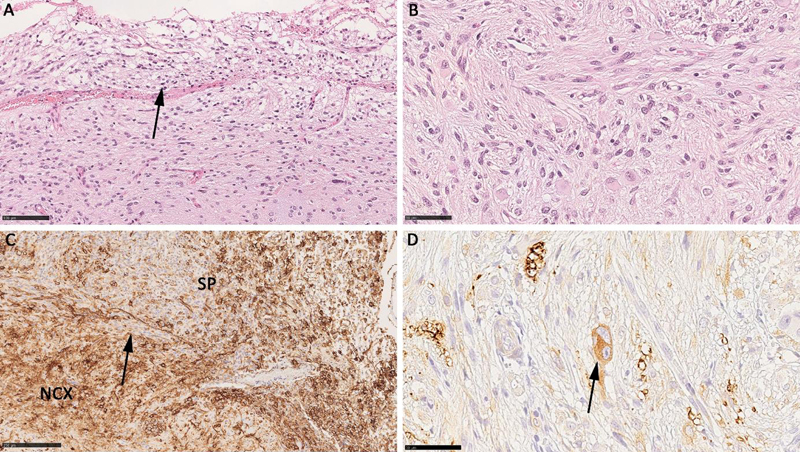
Figure 2: Histopathology findings in a PTPN11 altered atypical ganglioglioma. A: Subpial growth (arrow) with large, pleomorphic and glio-neuronal phenotype shown in B. C: abundant CD34 immunoreactivity (SP – subpial region). D: the arrow points to a bi-nucleated neuron (MAP2 immunohistochemistry) confirming the diagnosis of ganglioglioma.
2.04
Free Neuropathol 3:20:23
Molecular refinement of pilocytic astrocytoma in adult patients
Helena Bode1,2, Catena Kresbach1,2,3, Dörthe Holdhof1,2, Mario M. Dorostkar4,5, Patrick N. Harter6, Jürgen Hench7, Stephan Frank7, Alicia Eckhardt1,2,8, Annika K. Wefers3, Sina Neyazi1,2, David Capper9,10, Michael Bockmayr1,2,11, Ulrich Schüller1,2,3
1 Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
2 Research Institute Children’s Cancer Center Hamburg, Hamburg, Deutschland
3 Institute of Neuropathology, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
4 Center for Neuropathology, Ludwig-Maximilians-University, Munich, Deutschland
5 German Center for Neurodegenerative Diseases, Munich, Deutschland
6 Institute of Neurology (Edinger Institut), University Hospital Frankfurt, Frankfurt, Deutschland
7 Division of Neuropathology, Institute of Medical Genetics and Pathology, University of Basel, Basel, Switzerland
8 Lab of Radiobiology & Experimental Radiation Oncology, Hubertus Wald Tumorzentrum – University Cancer Center Hamburg, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
9 German Cancer Consortium (DKTK), Partner Site Berlin, and German Cancer Research Center (DKFZ), Heidelberg, Deutschland
10 Department of Neuropathology, Corporate Member of Freie Universität Berlin, Charité, Universitätsmedizin Berlin and Humboldt-Universität zu Berlin, Berlin, Deutschland
11 Institute of Pathology, Corporate Member of Freie Universität Berlin, Charité, Universitätsmedizin Berlin and Humboldt-Universität zu Berlin, Berlin, Deutschland
Background: Pilocytic astrocytomas (PA) are the most common primary central nervous system neoplasms in children. The vast majority of cases harbor KIAA1549-BRAF fusions and usually go along with an excellent prognosis. In contrast, PA in adult patients are rare, lack KIAA1549-BRAF fusions in many cases, and demonstrate a more aggressive clinical course.
Purpose: This project aims at characterizing adult PA regarding their molecular profile and clinical course.
Methods: We identified 55 cases with a histological diagnosis of PA in adulthood (≥18 years). Molecular analyses of these cases included DNA methylation analysis, copy number profiling, and DNA sequencing for the most common mutations in the MAPK-pathway.
Results: The mean age of our cohort was 35 years. Tumors were located infratentorially (41%), supratentorially (41%), and spinally (18%). After performing global DNA methylation analyses and applying the DKFZ brain tumor classifier (v12.5), only 25% of these cases received a significant match to one of the reference methylation classes of PA (score ≥ 0.9). 20% matched to different entities, and 55% did not match to any brain tumor class. Furthermore, only 23% of the tumors exhibited the KIAA1549-BRAF fusion. Further analyses of tumors with a significant match to one of the three PA reference classes showed that adult patients mostly had supratentorial PA (LGG_PA_GG_ST, mean age: 20 years, n=45), while children had PA in midline structures (LGG_PA_MID, mean age: 9 years, n=51) or in the posterior fossa (LGG_PA_PF, mean age: 11 years, n=159, p<0.005). Among these tumors defined by DNA methylation, the typical KIAA1549-BRAF fusion was found in 94 % of pediatric tumors and only in 45 % of tumors occurring in adults.
Conclusions: In summary, according to DNA methylation profiling, a particularly high fraction of tumors histologically appearing as PA in adult patients do not match known reference cohorts of PA. Many tumors are even reflecting other tumor entities, indicating ambiguous histological features. Furthermore, even in cases that significantly match to PA regarding DNA methylation, the distribution of genetic drivers differs from their pediatric counterparts.
2.05
Free Neuropathol 3:20:25
Exploration of cellular origins and therapeutic targets by modeling high grade pediatric glioma of the MYCN subclass in mice
Melanie Schoof1,2, Shweta Godbole3, Carolin Walter4,5, Matthias Dottermusch3,6, Thomas Albert5, Annika Ballast5, Carolin Göbel1,2, Sina Neyazi1,2, Dörthe Holdhof1,2, Catena Kresbach1,6, Gefion Dorothea Epplen1, Mirjam Blattner-Johnson7,8, Franziska Modemann9,10, Ann-Kristin Afflerbach1,2, Alicia Eckhardt1,11, Vanessa Thaden1, Nina Struve11,12, David T. W. Jones7,8, Kornelius Kerl5, Julia Neumann3,6, Ulrich Schüller1,2,6
1 Research Institute Children’s Cancer Center Hamburg, Hamburg, Germany
2 Department of Pediatric Hematology and Oncology, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
3 Center for Molecular Neurobiology (ZMNH), University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
4 Institute of Medical Informatics, University of Muenster, Muenster, Deutschland
5 Department of Pediatric Hematology and Oncology, University Children’s Hospital Münster, Muenster, Deutschland
6 Institute of Neuropathology, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
7 Hopp Children's Cancer Center (KiTZ), Heidelberg, Deutschland
8 Pediatric Glioma Research Group, German Cancer Research Center (DKFZ), Heidelberg, Deutschland
9 Department of Oncology, Hematology and Bone Marrow Transplantation with Division of Pneumology, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
10 Mildred Scheel Cancer Career Center, University Cancer Center Hamburg, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
11 Department of Radiotherapy, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
12 Mildred Scheel Cancer Career Center HaTriCS4, University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
Pediatric gliomas of the MYCN subclass, a recently described highly aggressive brain tumor entity, frequently carry amplifications of MYCN and mutations in TP53. These tumors present with a median age of 8 years and a median overall survival of only 14 months. Better treatment options are urgently needed, as the current treatment is ineffective and causes severe side effects. Here, we describe the generation of a novel mouse model, which can be used for preclinical research. We bred hGFAP-cre::TP53Fl/Fl::lsl-MYCN mice, which develop large forebrain tumors with 100 % penetrance within the first 80 days of life. The murine tumors show a high similarity with human tumors in histology, gene expression, and global DNA methylation pattern. Single-cell gene expression analyzes of these tumors revealed a large intratumoral cell heterogeneity and, due to the similarity of the tumor cells with oligodendrocytes in different developmental stages, suggests a glial origin of these tumors. Additionally, we tested the preclinical potential of our mouse model by showing sensitivity of mouse and human tumor cells to AURKA inhibition in vitro. We believe that further characterization and utilization of the model will pave the way to improved treatment strategies for patients with these highly aggressive tumors.
2.06
Free Neuropathol 3:20:27
The genomic and transcriptional landscape of primary central nervous system lymphoma
Josefine Radke1, Naveed Ishaque2, Reiner Siebert3, Stefan Wiemann4, Frank Heppner5
1 Universität Greifswald, Pathologie, Greifswald, Deutschland
2 Berlin Institute of Health (BIH), Digital Health Center, Berlin, Deutschland
3 Ulm University & Ulm University Medical Center, Human Genetics, Ulm, Deutschland
4 Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Deutschland
5 Charité, Neuropathology, Berlin, Deutschland
Background: Primary lymphomas of the central nervous system (PCNSL) are mainly diffuse large B-cell lymphomas (DLBCLs) confined to the central nervous system (CNS). Despite extensive research, the molecular alterations leading to PCNSL have not been fully elucidated.
Aims: In order to provide a comprehensive description of the genomic and transcriptional landscape of PCNSL, we here performed whole-genome and transcriptome sequencing and integrative analysis of 51 lymphomas presenting in the CNS, including 42 EBV-negative PCNSL, 6 secondary CNS lymphomas (SCNSL) and 3 EBV+ CNSL and matched controls. The results were compared to an independent validation cohort of 31 FFPE CNSL specimens (PCNSL, n = 19; SCNSL, n = 9; EBV+ CNSL, n = 3) as well as 39 FL and 36 systemic DLBCL cases outside the CNS.
Results: Somatic genomic alterations in PCNSL mainly affect the JAK-STAT, NFkB, and B-cell receptor signaling pathways, with hallmark recurrent mutations including MYD88 L265P (67%) and CD79B (63%), CDKN2A deletions (83%) and also non-coding RNA genes such as MALAT1 (70%), NEAT (60%), and MIR142 (80%). Kataegis events, which affected 15 of 50 identified driver genes and 21 of the top 50 mutated ncRNAs, played a decisive role in shaping the mutational repertoire of PCNSL. Compared to systemic DLBCL, PCNSLs exhibited significantly more focal deletions in 6p21 targeting the HLA-D locus that encodes for MHC class II molecules as a potential mechanism of immune evasion. Mutational signatures correlating with DNA replication and mitosis (SBS1, ID1 and ID2) were significantly enriched in PCNSL (SBS1: p = 0.0027, ID1/ID2: p < 1x10-4). Furthermore, TERT gene expression was significantly higher in PCNSL compared to ABC-DLBCL (p = 0.027). Although PCNSL share many genetic alterations with systemic ABC-DLBCL in the same signaling pathways, transcriptome analysis clearly distinguished both into distinct molecular subtypes. EBV+ CNSL cases may be distinguished by lack of recurrent mutational hotspots apart from IG and HLA-DRB loci.
Conclusion: We show that PCNSL can be clearly distinguished from DLBCL, having distinct expression profiles, IG expression and translocation patterns, as well as specific combinations of genetic alterations.
2.07
Free Neuropathol 3:20:28
Molecular mechanisms of therapy resistance in malignant melanoma brain metastasis
Elisa Schumann1, Randi Koll2, Julia Onken2, Karsten Jürchott2, Torben Redmer3, Josefine Radke4
1 Charité - Universitätsmedizin Berlin, Institut für Neuropathologie, Berlin, Deutschland
2 Charité - Universitätsmedizin Berlin, Berlin, Deutschland
3 Veterinärmedizinische Universität Wien, Wien, Austria
4 Universität Greifswald, Greifswald, Deutschland
Background: Malignant melanoma (MM) is among the tumor entities with the highest potential to spread to the CNS. About 45% of MM patients suffer from brain metastasis likely proceeding continuously during the course of disease. Genetically and molecularly distinct subclones lead to tumor heterogeneity which is followed by therapy resistance and poor prognosis. Previous studies suggested increased metastatic potential to and within the brain under BRAF inhibitor (BRAFi) therapy, which is caused by upregulation of a subset of molecular drivers controlling migratory and invasion such as the nerve growth factor receptor CD271/NGFR.
Aims: To gain insight into the molecular features of migration and invasion of patient derived cell lines from MM brain metastases (BM) that were therapy-responsive or therapy-resistant to BRAFi, radiotherapy and immune checkpoint inhibitors.
Methods: We generated patient derived cell lines from MM BM (n = 9) and performed DNA-sequencing (n = 5) and transcriptome analyses (n = 2) of cell lines and concordant tumors (n = 2). Furthermore, we used the Incucyte® Live-Cell Analysis to perform high throughput scratch wound assays with patient derived cell lines, which were genetically modified leading to overexpression or downregulation of NGFR.
Results: Transcriptome profiling of BRAFi resistant MM BM revealed that the invasive potential increased during disease progression. This process was accompanied by upregulation of NGFR expression. Moreover, it was preserved in patient derived cells lines, which demonstrated significantly higher potential of two-dimensional in vitro migration (90% vs. 76% after 100 hours). Furthermore, CD271 knockdown was associated with loss-of-expression of several genes involved in migration and invasion.
Conclusions: Brain metastases are the major cause of death in metastasized MM. Probably, BM emerge and progress by the concerted interaction of several molecular programs that are triggered by cells of the tumor microenvironment and/or in response to therapeutic interventions. Our study provides a longitudinal perspective on the progression of brain metastasis and their mechanisms leading to therapy resistance.
2.08
Free Neuropathol 3:20:29
A peripheral nerve sheath tumor syndrome caused by postzygotic ERBB2 mutations
Michael Ronellenfitsch1,2,3,4, Isabel Gugel5, Dusica Babovic-Vuksanovic6, Maximilian Rauch2,7, Jens Schittenhelm5, Martin U. Schuhmann5, Silvia Hofer8, Martina Kirchner9, Gerhard Marquardt10, Rouzbeh Banan911, Benedikt Sauer1,3, Ulrich Schüller12,13,14, Werner Paulus15, Matthias Meinhardt16, Tareq Juratli16, Albrecht Stenzinger9, Stefan Fröhling17,18, Eric Legius19, Andreas von Deimling9,11, Felix Sahm9,11, Joachim P. Steinbach1,2,3,4, Patrick Harter2,3,10, Victor-Felix Mautner12, David Reuss11,20
1 Dr. Senckenberg Institute of Neurooncology, Frankfurt, Deutschland
2 University Cancer Center (UCT) Frankfurt, Frankfurt, Deutschland
3 German Cancer Consortium (DKTK), Frankfurt, Deutschland
4 Frankfurt Cancer Institute, Frankfurt, Deutschland
5 University Hospital Tübingen, Tübingen, Deutschland
6 Mayo Clinic College of Medicine, Rochester, United States
7 Goethe University Hospital, Frankfurt, Deutschland
8 University Hospital and University of Zurich, Zürich, Switzerland
9 Heidelberg University Hospital, Heidelberg, Deutschland
10 University Hospital Frankfurt, Frankfurt, Deutschland
11 DKFZ, Heidelberg, Deutschland
12 University Hospital Hamburg-Eppendorf, Hamburg, Deutschland
13 University Medical Center Hamburg-Eppendorf, Hamburg, Deutschland
14 Research Institute Children's Cancer Center Hamburg, Hamburg, Deutschland
15 University Hospital Münster, Münster, Deutschland
16 University Hospital Carl Gustav Carus, Dresden, Deutschland
17 NCT Heidelberg and DKFZ, Heidelberg, Deutschland
18 DKTK, Heidelberg, Deutschland
19 KU Leuven and University Hospital, Leuven, Belgium
20 Universitätsklinikum Heidelberg, Neuropathologie, Heidelberg, Deutschland
Introduction: Peripheral nerve sheath tumors (NST) are common manifestations of different tumor syndromes within the neurofibromatosis spectrum, comprised of NF1, NF2 and schwannomatosis. These are caused by inactivating germline mutations in the NF1, NF2, SMARCB1 or LZTR1 tumor suppressor genes respectively. Neurofibromas are closely associated with NF1 and schwannomas occur in both NF2 and schwannomatosis. Occurrence of neurofibroma/schwannoma hybrid tumors is reported in all these syndromes. We recently described ERBB2 mutations in a significant portion of neurofibroma/schwannoma hybrid nerve sheath tumors. Based on clinical criteria, these cases resembled schwannomatosis. However, the somatic genetic profile of the tumors was distinct. Additionally, features untypical of schwannomatosis were present, resembling previously published descriptions of four patients with distinctive but unclassifiable clinical and pathological findings.
Objectives: The aim of the study was the clinicopathological and molecular characterization of ERBB2-mutant peripheral nerve sheath tumors.
Methods: Tumors were evaluated by histology. Next generation sequencing (HD-Panel or Whole exome sequencing) was used to determine the presence of an ERBB2 mutation in at least one tumor of every patient. Pyro-sequencing was used to verify ERBB2 mutations and to determine their presence in additional tumors from a given patient. 850k methylation profiling was used for additional characterizations.
Results: We identified 13 non-related patients with ERBB2 mutant NST, including all 4 previously published patients with an unclassified syndrome. All but two patients were females and tumors developed slowly during adulthood. All patients had multiple NST, which were restricted to a specific anatomic region in several patients while a more widespread distribution of tumors was present in others. The histology showed quite distinctive features within the spectrum of hybrid nerve sheath tumors. Strikingly, in all patients, tumors from distinct anatomic locations harbored the very same activating ERBB2 mutation. The median mutant allele frequency of 12% was comparatively low (range 4%-23%) suggesting that only a subpopulation of cells harbored the mutation. No other candidate driver alteration was found by NGS panel or WES and RNA-sequencing. DNA methylation profiling provided evidence for a distinctive epigenetic profile of ERBB2-mutant NSTs. No chromosomal copy number alterations were detectable. Ongoing molecular analyses will provide additional insight in the pathogenesis of ERBB2-mutant NSTs.
Conclusion: ERBB2 mutations in NST do not occur as isolated somatic events in sporadic tumorigenesis or in the setting of NF1, NF2 or schwannomatosis but represent manifestations of a distinct tumor syndrome most likely caused by postzygotic ERBB2-mosaicism. Diagnosis of ERBB2-mutant NST is of high clinical relevance due to the availability of specific ERBB2 inhibitors and preliminary evidence of their effectiveness. Histology is sufficiently specific for screening purposes but molecular analyses with highly sensitive methods like deep coverage NGS are mandatory for the definitive diagnosis.
2.09
Free Neuropathol 3:20:31
CNS-tumor patients within the IMPRESS-Norway trial: First year experiences
Pitt Niehusmann1,2, Hege G Russnes1,3,4, Katarina Puco2, Åsmund Flobak5,6, Eli Sihn S. Steinskog7, Åse Haug7, Sigmund Brabrand2,8, Egil S. Blix9,10, Anne J Skjulsvik5,11, Ragnhild M Wold12, Henning Leske1, Hrvoje Miletic13,14, Petter Brandal8,15, Gro L. Fagereng16, Kjetil Taskén3,17, Åslaug Helland3,4,8
1 Oslo University Hospital, Department of Pathology, Oslo, Norway
2 Oslo University Hospital, Division for Cancer Medicine, Oslo, Norway
3 University of Oslo, Institute of Clinical Medicine, Oslo, Norway
4 Oslo University Hospital, Department of Cancer Genetics, Institute for Cancer Research, Oslo, Norway
5 Norwegian University of Science and Technology, Department of Clinical and Molecular Medicine, Trondheim, Norway
6 St. Olav University Hospital, The Cancer Clinic, Trondheim, Norway
7 Haukeland University Hospital, Department of Oncology, Bergen, Norway
8 Oslo University Hospital, Department of Oncology, Oslo, Norway
9 UiT - The Arctic University of Norway, Institute of Clinical Medicine, Tromsø, Norway
10 University Hospital of North Norway, Department of Oncology, Tromsø, Norway
11 St. Olav University Hospital, Department of Pathology, Trondheim, Norway
12 University Hospital of North Norway, Department of Pathology, Tromsø, Norway
13 University of Bergen, Department of Biomedicine, Bergen, Norway
14 Haukeland University Hospital, Department of Pathology, Bergen, Norway
15 Oslo University Hospital, Section for Cancer Cytogenetics, Institute for Cancer Genetics and Informatics, Oslo, Norway
16 Oslo University Hospital, Institute for Cancer Research, Oslo, Norway
17 Oslo University Hospital, Department of Cancer Immunology, Institute for Cancer Research, Oslo, Norway
Background: IMPRESS-Norway is a nation-wide precision medicine trial for cancer patients in Norway that launched April 1st 2021. In this investigator-initiated, prospective, open-label, non-randomized combined basket- and umbrella-trial, patients are enrolled into multiple parallel treatment cohorts. Patients with progressive cancer disease, including primary CNS-neoplasms, with no further standard therapy to offer, are eligible. All drugs available in IMPRESS-Norway are regulatory approved. Currently, five different pharmaceutical companies provide 16 drugs, and we are in process to acquire eight additional drugs for patients in this study.
Methods: Comprehensive genomic profiling (gene-panel analysis of >500 genes) is performed as part of the Norwegian public health care system. Patients consenting to the IMPRESS-Norway profiling phase contribute their clinical and molecular data for research and are screened for cell-free circulating tumor DNA. Patients with identified biomarkers matching available drugs are referred by the national molecular tumor board for inclusion in the IMPRESS-Norway treatment phase. These patients will have extensive biobanking as well as whole genome molecular profiling of their tumors before and during treatment. In the IMPRESS-Norway treatment phase, each cohort is defined by the patients’ tumor type, molecular profile of the tumor, and study drug. Treatment outcome in each cohort is monitored using a Simon two-stage-like ‘admissible’ monitoring plan to identify evidence of clinical activity. The primary objective in the study is clinical benefit of treatment at 16 weeks of treatment; defined as complete response, partial response, or stable disease. Here, we report on patients with CNS-neoplasms included in the IMPRESS-Norway profiling and treatment phases.
Results: As of April 30th, 2022, twenty-four patients with CNS-neoplasms had been included in the molecular profiling phase of IMPRESS-Norway and 22 had completed evaluation at the molecular tumor board (see Table 1). Tumor mutation burden (TMB) in CNS-tumor tissue samples ranged from 1.6-250 somatic mutations per megabase (mut/Mb; median=4.7, n=23). In liquid biopsies, blood TMB ranged from 0-6 mut/Mb (median=0; n=21), indicating a limited efficacy of this analysis in CNS-tumor patients. In five of the 22 patients with completed evaluation, we identified biomarker, which allowed allocation to an IMPRESS-Norway treatment-cohort (ratio of CNS-patients with targetable biomarker was similar to the overall inclusion of patients into treatment cohorts, 67/295). One glioblastoma patient showed complete response according to RANO at 39 weeks.
Table 1
| Diagnosis | Treatment cohort |
| Glioblastoma, IDH-wildtype (n=13) | n=3 |
| Astrocytoma, IDH-mutant (n=4) | n=0 |
| Anaplastic meningioma (n=1) | n=0 |
| Atypical meningioma (n=1) | n=0 |
| Diffuse midline glioma, H3 K27-altered (n=1) | n=1 |
| High-grade astrocytoma with piloid features (n=1) | n=1 |
| Myxopapillary ependymoma (n=2) | n=0 |
| Supratentorial ependymoma (n=1) | n=0 |
Due to increasing test capacity, we anticipate to double the number of included CNS-tumor patients within the next 6 months. Whole genome sequencing data from patients included into the treatment cohorts are obtained successively.
Conclusion: Patients with advanced cancer progressing on standard treatment are referred to treatment in IMPRESS-Norway after advanced molecular diagnostics. Molecular alterations indicating benefit of drugs currently available in the study are detected in a reasonable number of patients with CNS-neoplasms.
3. Neurodegeneration
3.01
Free Neuropathol 3:20:33
CNN-supported quantification of fat compartments at abdominal MRI applied to ALS patients
Ina Vernikouskaya1, Hans-Peter Müller2, Dominik Felbel1, Francesco Roselli2, Albert Christian Ludolph2, Volker Rasche1, Jan Kassubek2
1 Ulm University Medical Center, Internal Medicine II, Ulm, Deutschland
2 University of Ulm, Neurology, Ulm, Deutschland
Background: Amyotrophic lateral sclerosis (ALS) is the most frequent adult onset neurodegenerative motor neuron disease characterized by catabolism1, and patients begin to lose weight more than 10 years before the onset of motor symptoms2. ALS patients have been shown to display an expanded ratio between visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT)3. Accurate segmentation of body fat compartments from MRI is, however, a challenging task due to the limited reproducibility of semi-manual delineations and artifacts. Concerning organ segmentation, learning-based algorithms and especially convolutional neural networks (CNN) have been proven to outperform traditional methods in speed and reproducibility.
Aims: The aim of this study was to automate the discrimination of abdominal body fat compartments into SAT and VAT from T1-weighted MRI using deep CNN and to quantify the fat ratio in patients with ALS as compared to the control cohort.
Question: May CNN-supported segmentation of body fat compartments serve for unbiased analysis of the VAT/SAT ratio parameter as a potential biological marker?
Methods: 74 ALS patients (age 60 ± 12, m/f 50/24) and 81 healthy subjects (56 ± 15, 42/39) underwent MRI examination with multi-slice T1-weighted spin-echo sequence. All available data were split in training (50 %), validation (6 %), and test (44 %) data, based on age and BMI strata. Semi-automatic segmentation of subcutaneous and visceral fat was performed with an established reference method using software package ATLAS4. The obtained SAT/VAT masks were used for training of the CNN of U-Net like architecture. Performance of the segmentation using CNN was evaluated in terms of dice coefficients. Volumetric computation of segmented SAT and VAT for all test objects was performed with reference and CNN-based methods and compared by Pearson correlation. VAT/SAT ratio was assessed.
Results: The dice coefficients between the CNN-supported and reference segmentations comprised 0.87 ± 0.04 for SAT and 0.64 ± 0.17 for VAT in the control group and 0.87 ± 0.08 for SAT and 0.68 ± 0.15 for VAT in the ALS group. A significant linear correlation between the CNN predicted and reference method with Pearson coefficients 0.992 in controls and 0.977 in ALS patients was observed for SAT, whereas lower Pearson coefficients 0.653 in controls and 0.814 in ALS patients were obtained for VAT. Significant difference for the VAT/SAT ratio was observed when comparing ALS patients versus healthy subjects with the p-value of 0.002. Figure 1 shows the SAT and VAT segmentation results in healthy controls and ALS patients.
Conclusions: The obtained results in the T1-weighted MRI data in the ALS patient cohort could reproduce the results of a reference technique in a user-independent manner with high accuracy. CNN-supported quantification of VAT/SAT ratio might serve as a biological marker in ALS body composition assessment, potentially as a secondary read-out for clinical trials.
References:
1. Dupuis et al, Lancet Neurol. 2011
2. Peter et al, Eur J Epidemiol. 2017
3. Lindauer et al, PLoS One. 2013
4. Müller et al, NMR Biomed. 2011
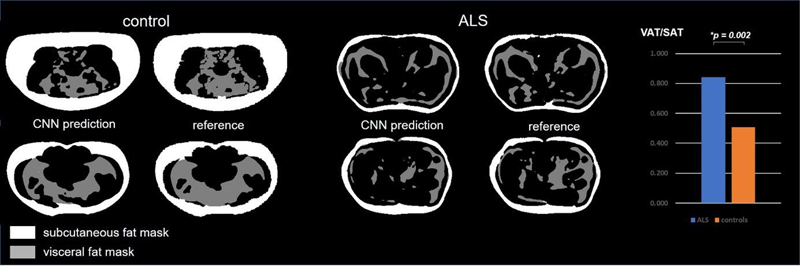
Figure 1: Comparison of CCN-based segmentation vs. reference segmentation and VAT/SAT ratio plot in healthy controls and ALS patients.
3.02
Free Neuropathol 3:20:35
Neurodegenerative iron storage disease (neuroferritinopathy) caused by a novel frameshift mutation in the ferritin heavy chain gene (FTH1 c.341-342del)
Vincent Umathum1,2, Daniel Amsel1, Christina Becker3, Corinne Kasan3, Andrea May4, Klaus Nehmer4, Andreas Günther4,5, Carmen Selignow1, Anna Nishimura1, Ioannis Alexopoulos6, Attila Németh1, Nadja Ritschel1, Axel Weber7, Till Acker1, Anne Schänzer1
1 Institut für Neuropathologie, Justus-Liebig-Universität, Gießen, Deutschland
2 Institut für Pathologie und Molekularpathologie, Bundeswehrkrankenhaus Ulm, Ulm, Deutschland
3 Institut für Pathologie, Zytologie und Molekularpathologie MVZ, Wetzlar, Deutschland
4 Pneumologische Klinik, Agaplesion Evangelisches Krankenhaus, Gießen, Deutschland
5 Zentrum für Interstitielle und Seltene Lungenerkrankungen, Justus-Liebig-Universität, Gießen, Deutschland
6 Institute for Lung Health, Justus-Liebig-Universität, Gießen, Deutschland
7 Institut für Humangentik, Justus-Liebig-Universität, Gießen, Deutschland
Introduction: Neuroferritinopathy (NF) is a rare hereditary neurodegenerative disorder associated with increased iron deposition in the brain and extracerebral organs such as the kidney, liver, skin and skeletal muscle. The ferritin complex consists of 24 subunits of ferritin light and heavy chains and converts free iron into a non-redox active storage form. Clinically, the focus is on Chorea-Huntington-like movement disorders. So far, only mutations in the ferritin light chain gene (FTL) have been described in NF.
Material and methods: A 78-year-old female patient died of covid-19-associated pneumonia and was autopsied. Standard staining (HE, Prussian Blue), immunohistochemical and immunofluorescence staining as well as electron microscopic analyses were performed on paraffin-embedded formalin-fixed (FFPE) tissue of the patient from different brain regions as well as heart, lung, liver and kidney. Whole slide images scans were done by Hamamatsu NanoZoomer S360 and evaluated morphometrically with QPath. Whole-exome sequencing (WES) was done from FFPE material of the basal ganglia.
Results: Macroscopically, no pathology was found in the brain. Microscopically, numerous inclusion bodies (IB) were seen in the brain and sporadically in the liver and kidney. The IB were homogeneously sharply defined on HE stains and up to 13 µm in size (normal nuclear diameter: approx. 5-6 µm), with strong Fe3+ deposits in the Prussian blue staining.
Ultrastructurally, the IB showed intranuclear, fine granular aggregates with lateralisation of the chromatin to the inner side of the membrane. Immunohistochemistry for FTL and ferritin heavy chain protein (FTH) showed clear nuclear expression in the IB. In contrast, the cells in the control tissue had predominantly perinuclear, cytoplasmic expression. Quantitative evaluation showed increased FTH expression in the patient: frontal cortex (CF): 0.2%, occipital cortex (CO): 0.3%, hippocampus: 0.08%, basal ganglia (BG): 0.7%, dentate nucleus (DN): 1.7% compared to control tissue (CF: 0.06% CO: 0.04% Hippocampus: 0.04% BG: 0.08% DN: 0.4%. The ratio of FTH expression (compared to control tissue) was highest in the BG (1:8.4), followed by the CO (1:8.3) and lowest in the hippocampus (1:2.1). WES revealed a previously undescribed variant (double deletion) in the ferritin heavy chain gene (FTH1 c.341-342del). Wild-type sequence in FTL.
Discussion: The present study describes for the first time a patient with NF caused by a previously undescribed mutation in the FTH1 gene with presentation of numerous IB in the brain and sporadically in extracerebral tissue. These results suggest that the function of the ferritin complex can be disturbed not only by an FTL- but also by an FTH1-mutation, leading to pathological deposition of ferritin complexes. With extended analyses, it could be shown that the IB correspond to enlarged cell nuclei with intranuclear ferritin accumulations. Furthermore, a high variability in distribution of the IB in different brain regions was found.
Summary: Mutation in FTH1 (c.341-342del) is associated with a rare neurodegenerative disease with increased intranuclear iron deposition mainly in the BG and DN, possibly showing a similar pathomechanism to the known FTL-mutation. If NF is clinically suspected, mutations should therefore be investigated not only in the FTL-gene but also in the FTH1-gene.
3.03
Free Neuropathol 3:20:37
The contribution of LATE-NC to neuron loss, granulovacuolar
degeneration and dementia in Alzheimer’s disease
Dietmar Thal1,2,3, Klara Gawor1,3, Evelien Van Schoor1,3,4, Sebastiaan Moonen1,3,4, Jolien Schaeverbeke1,3,4, Rik Vandenberghe3,4,5, Mathieu Vandenbulcke3,4,6, Christine A. F. von Arnim7,8, Marta Koper1,3,4, Sandra Tomé1,3
1 KU-Leuven, Department of Imaging and Pathology, Laboratory for Neuropathology, Leuven, Belgium
2 UZ Leuven, Department of Pathology, Leuven, Belgium
3 Leuven Brain Institute, Leuven, Belgium
4 KU Leuven, Department of Neuroscience, Leuven, Belgium
5 UZ Leuven, Department of Neurology, Leuven, Belgium
6 UZ Leuven, Department of Psychiatry, Leuven, Belgium
7 Göttingen University, Department of Geriatrics, Göttingen, Deutschland
8 Ulm University, Neurology, Ulm, Deutschland
Background: TDP-43 pathology in Alzheimer’s disease (AD) is currently considered as a co-pathology belonging to the spectrum of limbic-predominant, age-associated TDP-43 encephalopathy (LATE). AD cases with TDP-43 pathology have greater medial temporal lobe atrophy and cognitive decline compared to AD cases without TDP-43. Recently, we showed that the accumulation of the necrosome (executer complex of necroptosis which is a programmed from of necrosis) in granulovacuolar degeneration (GVD) in AD is associated with neuron loss.
Aims: Here, we aim to clarify whether necroptosis in AD is related to TDP-43 pathology, i.e., LATE neuropathological changes (LATE-NC) and indicates a contribution of LATE-NC to the degeneration/ death of neurons in AD. By doing so, we will determine the impact of LATE-NC on AD-NC, neuron loss, especially necroptosis, and cognitive decline.
Research question: Does TDP-43 pathology in LATE contribute to neurodegeneration via necrosome accumulation and necroptosis in AD and, if so, is this related to tau pathology.
Methods: We used 234 human post-mortem brains from 89 non-AD controls, 82 p-preAD and 63 AD cases. We determined amyloid-β (Aβ) (MTL = medial temporal lobe-based) phases, Braak neurofibrillary tangle (NFT) stages, LATE-NC stages and GVD stages, which described the anatomical spread of GVD bodies, and the frequency of GVD-affected neurons in a given region. From these, 66 cases were assessed for neuronal density in the CA1 subfield of the hippocampal formation and 186 were retrospectively assessed for clinical dementia rating (CDR) scores. In a subset of 27 cases covering 9 non-AD controls (without TDP-43 pathology), 8 symptomatic AD cases without TDP-43 pathology (ADTDP-) and 10 symptomatic AD cases with TDP-43 proteinopathy (i.e.: LATE-NC; ADTDP+), we quantified neuronal density, severity of pTDP-43, pMLKL (a component of the activated necrosome) and pTau pathology.
Results: We found that LATE-NC and Braak NFT stages contribute independently to GVD severity when being covariates the same model term (p < 0.001). LATE-NC and Braak NFT stages were associated with neuronal density in the hippocampus (p = 0.022 and p = 0.004, respectively). Braak NFT stage alone showed also an association with neuronal density (p < 0.001). Partial correlation analysis corroborated these data and revealed that Braak NFT and LATE-NC stage are significantly correlated with GVD stage and CDR score. In the 27 cases used for quantitative assessments, the absence of pTDP-43 was associated with less neuronal loss in ADTDP- cases compared to ADTDP+ (p = 0.0204). ADTDP+ cases exhibited significantly more pMLKL-positive neurons, when compared to ADTDP- cases (p = 0.025) and non-diseased controls (p < 0.0001). Consistently, ADTDP+ cases also showed enhanced numbers of pTau exhibiting neurons compared to ADTDP- (p = 0.045) and controls (p < 0.0001).
Conclusions: Thus, LATE-NC contributes independently to neuronal loss and GVD severity. Further, these data highlight the impact of cytoplasmic TDP-43 aggregation on pTau pathology as well as on necroptosis activation in AD.
Support: FWO.
3.04
Free Neuropathol 3:20:39
The role of C3 inhibition in an iPSC NMJ model of neuroinflam-mation
Scott Baver1, Virginia Smith2, Yan LI1, David Eyerman1, Ashley Robertson2, Leticia Lenkiu2, Heather Cannon2, Daisy Martinez2, Hannah Hanson2, James Hesperos2
1 Apellis, Waltham, United States
2 Hesperos, Orlando, United States
Background: The complement cascade is a critical component of the immune system, and dysfunction of complement has been implicated in ALS. Components of the complement cascade, including C3, are reported to be deposited on neuromuscular junctions (NMJs) of muscle biopsies in people with ALS.
Objectives: Evaluate the effect of complement C3 inhibition on NMJ function in response to inflammatory stimulations in a human iPSC-derived NMJ model.
Question: While the therapeutic potential of complement system modulation has been explored via use of embryonic knockout animals in ALS models, its effects in clinically relevant human cell-based models are unknown.
Methods: NMJ systems were established by plating human iPSC-derived motoneurons, skeletal myoblasts, Schwann cells, microglia, and activated or inactivated THP monocytes in a compartmentalized co-culture system. Activated and inactivated monocytes were plated at ratios of 4:1-1:50 (vs. skeletal myoblasts). ALS model systems were created using SOD-1 (E100G) or TDP-43 iPSC-derived cells compared to control NMJs established from wild-type iPSC-derived cells. To determine the role of C3 on NMJ function, the C3 inhibitor APL-2 (50 µg/mL) and human complement serum (0.05%) were acutely dosed for 3 hours. Complement C3 expression was assessed by immunocytochemistry, and NMJ number and fidelity were calculated by assessing the number of functional myotubes under indirect stimulation and the ratio of number of successful contractions to number of pulses at a given frequency, respectively. All experiments were replicated twice in triplicate.
Results: Functional NMJ systems were assessed with the addition of various ratios of activated or inactivated monocytes to skeletal muscle-side of the culture chamber. Addition of activated monocytes resulted in reduced NMJ number and function. In addition, while C3 expression was observed with THP-monocytes, activation of M1 macrophages increased C3 activity. Human complement serum potentiated the effects of M1 macrophages; further decreasing NMJ numbers and reducing NMJ fidelity. Acute treatment with APL-2 attenuated these effects. Compared to wild-type, SOD-1 and TDP-43 NMJ systems reduced NMJ number and fidelity. The reduction in SOD-1 NMJ number and fidelity was greater than that of TDP-43 NMJ system.
Conclusions: These data demonstrate that modulating C3 with APL-2 in the presence of an inflammatory NMJ environment could improve overall function of the NMJ in related disease states.
3.05
Free Neuropathol 3:20:40
Fast-track procedure for the neuropathological assessment of neurodegenerative diseases
Benjamin Englert1,2,3, Viktoria Ruf1, Jochen Herms1,2,3
1 Ludwig-Maximilians University, Center for Neuropathology and Prion research, Munich, Deutschland
2 German Center for Neurodegenerative Diseases (DZNE), Munich, Deutschland
3 Munich Cluster for Systems Neurology (SyNergy), Munich, Deutschland
Background: In order to exclude Creutzfeldt-Jakob disease we have established a fast-track assessment of all brains that we receive at the Neurobiobank Munich.
Aims and questions: We aim to evaluate the concordance between fast-track working diagnosis and the full histological work-up of the brain of patients clinically diagnosed with a neurodegenerative disease.
Methods: Two predefined, easily accessible brain regions (frontal superior gyrus and cerebellar hemisphere) were sampled, fixed in formalin, treated with formic acid and embedded together in one paraffin block. H&E stain and six immunostains (antibodies against prion protein, α-Synuclein, β-Amyloid, phospho-Tau, phospho-TDP43, p62) were performed. 133 cases were analysed. Diseases in which a diagnosis cannot be made by analysing these two brain regions like pure amyotrophic lateral sclerosis and Huntington disease have been excluded from this analysis.
Results: In 96.2% of cases the fast-track neuropathological diagnosis was confirmed by the conventional pathological work-up of the entire brain. Only in four cases evaluation of additional brain regions was necessary to make a conclusive histopathological diagnosis. Only in one case in our study (of an unusual tauopathy) the fast-track diagnosis needed to be corrected.
The suspected clinical diagnosis was neuropathologically confirmed by fast-track histology in only 60.2 % of cases. In 39.8 % the clinical diagnosis was either different or ambiguous.
Conclusion: Our study shows that histological and immunohistochemical screening of two selected brain regions (superior frontal gyrus and cerebellum) is indeed sufficient for establishing a reliable working diagnosis of patients with a neurodegenerative disease in over 96 % of cases. Given the rapid workflow, a prompt response from the neuropathology to clinicians may improve the accuracy of clinical diagnosis.
This procedure does not allow precise neuropathological disease staging and certain co-pathology cannot be fully appraised. Diseases like pure ALS or HD cannot be neuropathologically verified on these two brain regions and the contribution of vascular pathology to the clinical picture cannot be precisely assessed.
3.06
Free Neuropathol 3:20:41
Neurodegeneration in HSAN1 due to ATL1 (Gly66Gln) mutation is associated with defective ER- protein quality control and compromised autophagy
Istvan Katona1, Hülya-Sevcan Daimagüler2, Haihong Guo1, Priyanka Tripathi1, Antonio Sechi3, Alfred Yamoah1, Shelisa Tey1, Michael Schröder1, Jürgen Klingelhöfer4, Joachim Weis1, Anand Goswami1
1 Uniklinik RWTH Aachen, Institut für Neuropathologie, Aachen, Deutschland
2 Uniklinik Köln, Kinderklinik, Köln, Deutschland
3 Uniklinik RWTH Aachen, Institut für Zell- und Tumorbiologie, Aachen, Deutschland
4 Klinikum Chemnitz , Klinik für Neurologie, Chemnitz, Deutschland
Background: Atlastin-1 (ATL1) functions as a GTPase and is crucial for endoplasmic reticulum (ER) shaping and ER-microtubule interactions. Mutations in ATL1 have been reported to cause hereditary sensory and autonomic neuropathy type 1D (HSAN1D) as well as hereditary spastic paraplegia 3A (SPG3A).
ATL1 mutation have been linked to abnormal ER morphology; still the molecular pathomechanism of defective ER structures and their pathological consequences contributing to HSAN1D and SPG3A have not been investigated in detail so far.
Question: We asked if/how ATL1 participates in autophagy process.
Methods: We used biochemical and immunocytochemistry approaches followed by live cell imaging in cell culture models overexpressing normal and mutant ATL1 proteins. We corroborated our findings with comparative ultrastructural analysis on cell culture models and on biopsy samples.
Results: We observed that over-expression of mutant ATL1 (Gly66Gln) forms protease resistant large, globular ER associated aggregates in cell culture models, which further leads to ER stress and structural abnormalities of ER and associated compartments. Interestingly while endogenous ATL1 protein is degraded by ubiquitin proteasome system (UPS), mutant ATL1 impairs UPS, induces proteotoxicity and cell death. Autophagy, which activates as a compensatory mechanism, also compromises at multiple steps, probably due to deformities of ER and persistent proteotoxic stress. Extensive workup of skin, sural nerve and muscle biopsy material of a rare Gly66Gln HSAN1 patient revealed prominent loss of myelinated and unmyelinated sural nerve fibres, but only minor neurogenic muscular atrophy. Ultrastructural analysis on this biopsy revealed signs of altered autophagy in axons as well as prominent alterations of Schwann cell nuclei/nuclear envelope. In line with this, HSAN1 patient's fibroblasts showed similar defects.
Conclusions: Overall, our results support the notion that neurodegeneration in HSAN1 due to ATL1 mutation is closely linked with the deformed ER and associated functions including autophagy. Neurons and distal axons are particularly vulnerable to such pathomechanism, thus explaining the degenerative phenotype in HSAN 1 and related diseases.
3.07
Free Neuropathol 3:20:42
Single-Nucleus Chromatin Accessibility Profiling in Four-repeat Tauopathies
Viktoria Ruf1, Nils Briel1,2, Sigrun Roeber1, Janina Mielke1, Mario M Dorostkar1, Otto Windl1,2, Thomas Arzberger1,2,3, Felix L. Strübing1,2, Jochen Herms1,2,4
1 Zentrum für Neuropathologie und Prionforschung, München, Deutschland
2 Deutsches Zentrum für Neurodegenerative Erkrankungen, München, Deutschland
3 Klinik für Psychiatrie und Psychotherapie, München, Deutschland
4 Munich Cluster of Systems Neurology (SyNergy), München, Deutschland
Background: Progressive supranuclear Palsy (PSP) and Corticobasal degeneration (CBD) are sporadic neurodegenerative diseases characterized by aggregates of hyperphosphorylated four-repeat tau (4R-tau) in neurons, oligodendrocytes and astrocytes, where tufted astrocytes (TA) are a typical hallmark of PSP, whereas astrocytic plaques (AP) are pathognomonic for CBD. The molecular mechanisms underlying tau aggregation and neurodegeneration are largely unclear.
Aims and questions: To characterize chromatin accessibility profiles of PSP and CBD using ATAC-Seq (Assay for Transposase-Accessible Chromatin using Sequencing) to contribute to a better understanding of the underlying pathomechanisms of PSP and CBD.
Methods: ATAC-Seq was performed on 45,000 isolated single nuclei from the frontal cortex of 4 PSP and 4 CBD patients and 5 healthy controls. After preprocessing and quality control, gene accessibility (GA), gene ontology (GO) and transcription factor motif enrichment (TFME) analysis were conducted using SnapATAC. gchromVAR was applied to map genetic risk variants to peaks and Cicero/tradeSeq was used for pseudotime analysis and repeated TFME analysis.
Results: We found that GA of tauopathy-associated genes was substantially altered in the brains of PSP and CBD patients and could demonstrate that PSP- and FTD-associated genetic risk variants are particularly linked to astrocytic chromatin accessibility profiles. GO enrichment analysis of significantly different transcription factor motifs (TFM) in astrocytes identified numerous motifs belonging to the immediate early response (IER; e.g. FOS and JUN family) or intracellular homeostasis and protein degradation (e.g. MAF family and NFE2). Moreover, pathway analysis highlighted immunological terms related to both innate and acquired immunity. Pseudotime analysis of transcription factor motif enrichment (TFME) revealed for CBD a trajectory terminating in a population of CBD-derived astrocytes, whereas for PSP no such disease-defined cluster was apparent. Accordingly, a decrease was observed for TFs associated with early state astrocytic differentiation in CBD, while TFME of IER-related TFs considerably increased along the pseudotime axis.
Conclusions: Our study revealed genetic dysregulation affecting neurodegenerative, neuroinflammatory and degradation processes and particularly points to a major role of astrocytes in the pathogenesis of PSP and CBD. However, to understand the downstream mechanisms, further independent validation especially at the transcriptome and protein level will be necessary.
Github ressource: https://github.com/nes-b/snATAC-seq_psp_cbd
3.08
Free Neuropathol 3:20:43
Application of a human stem cell transplantation model of Alzheimer’s disease to examine disease-associated changes at a single cell level in vivo
Wenhui Qu1, Matti Lam2, Aayushi Mahajan3, Nelson Humala3, Osama Al Dalahmah1, Jason Mares2, Trang Nguyen1, Ismael Santa-Maria1, Andrew Sproul1, Markus Siegelin1, James Goldman1, Peter Canoll1, Vilas Menon2, Gunnar Hargus1
1 Columbia University Medical Center, Department of Pathology and Cell Biology, New York, United States
2 Columbia University Medical Center, Department of Neurology, New York, United States
3 Columbia University Medical Center, Department of Neurosurgery, New York, United States
Background: Alzheimer’s disease (AD) is the most common type of dementia and is characterized by widespread degeneration of the central nervous system with amyloid and tau pathology leading to severe impairment of learning and memory. Despite the high number of patients with AD, the molecular mechanisms leading to neurodegeneration are only partially understood and effective treatment options still do not exist.
Aims/Questions: Here, we applied a human stem cell model of AD to study disease-modifying roles of neural cells at a single cell level in vivo.
Methods: To this end, we differentiated induced pluripotent stem cells (iPSCs) with the familial AD-associated APPV717I mutation in the amyloid precursor protein as well as isogenic control cells into neural progenitor cells (NPCs) and neurons.
Results: APPV717I neurons showed reduced neurite outgrowth and demonstrated an increased susceptibility towards oxidative stress with changes in metabolic programs. We injected APPV717I and control NPCs into the brains of immunocompromised NSG mice resulting in neuronal grafts in both groups two months after injection with activation of microglial cells and presence of reactive astrocytes within and around the grafts. We then performed single nucleus RNA sequencing (snRNA-seq) on microdissected grafts to characterize gene expression and dysregulated pathways in APPV717I versus control neurons as well as in astroglial cells and microglia in response to APPV717I and control neurons at a single cell level.
Conclusions: Our findings show that iPSCs represent a powerful cell source to study mechanisms of disease development in vitro and in vivo. This stem cell model of AD could also be used as a cellular platform for high-throughput drug screening purposes to identify potential therapeutic targets in AD.
4. Neuroinflammation
4.01
Free Neuropathol 3:20:44
Pathological And Genetic Characterization Of JC Virus Encephalopathy With An Eleven-Year-Long Disease Course
Marco Mlynek1, Adriane Kuttlovci1, Marek Jauß2, Cornelia Tennstedt-Schenk3, Lidia Stork1, Christine Stadelmann1, Imke Metz1
1 Institut für Neuropathologie - Universitätsmedizin Göttingen, Göttingen, Deutschland
2 Ökumenisches Hainich Klinikum gGmbH, Mühlhausen/Thüringen, Deutschland
3 Institut für Pathologie, Mühlhausen/Thüringen, Deutschland
The JC polyomavirus (JCPyV) typically causes progressive multifocal leukoencephalopathy (PML) in immunocompromised humans. The pathology shows demyelinated white matter lesions with infection of glial cells, but very few infected cortical pyramidal neurons. In contrast, another JCPyV-associated disease is JC virus encephalopathy (JCVE), which is characterized by numerous infected neurons. While the non-coding control region of JCPyV in PML shows genetic rearrangements (so-called prototype), in JCVE the archetype virus is found, which is also present in healthy individuals. We provide a detailed clinical, (histo)pathological and genetic description of a patient with JCVE. We analyzed different lesion areas for viral infection and replication of glial and neuronal cells. In addition, demyelination, cell loss and axonal damage were investigated. We present a 54-year-old male patient with an exceptionally long disease course over eleven years who was diagnosed with JCVE. He developed slowly progressing myoclonia, numbness of the upper and lower extremities, pharmacoresistant seizures and a general cognitive decline. MRI showed a progressive atrophy of the cortex with cortical and subcortical parenchymal lesions. There was no clear evidence of immunosuppression. A variant of unclear significance in the interferon-induced with helicase C domain 1 (IFIH1) gene was found. He died due to status epilepticus and pneumonia. Macroscopy showed extensive cystic white matter lesions with preservation of the cortical ribbon (Figure 1A). Histology revealed in addition lesions with ongoing viral infection. Here, the number of infected neuronal and glial cells outnumbered clearly the number of replicating cells, suggesting a limited viral replication (Figure 1B). Nevertheless, a pronounced decrease in oligodendrocytes was observed, while no major loss of astrocytes and neurons was found. Juxtacortical lesions showed prominent axonal damage and loss. Late lesions were characterized by a necrotic-cystic tissue defect located primarily at the gray-white junction and later spreading throughout the white matter. Only few publications describe JCVE as a new entity of JC virus pathologies characterized by a prominent infection of neurons. What distinguishes our case from others is the extraordinarily long disease course without clear evidence of immunodeficiency. A variant of unclear significance in the IFIH1 gene was found. IFIH1 encodes the MDA5 protein (pattern-recognition-receptor), which is part of the innate immune system and is mainly responsible for the activation of the antiviral immune response such as the induction of interferon-1/-β/-α. This mutation potentially predisposed to the JCPyV infection. The slow disease progression in our patient is possibly due to relatively well-preserved immune function. The presence of a stable, archetype variant could also indicate a less productive viral variant. Our detailed histological description shows that infected cells clearly outnumbered replicating cells, leading to production of virus in only single cells. Despite numerous infected neurons, in the long run white matter lesions with a pronounced tissue destruction prevailed. Thus, destructive white matter lesions seem to be a pivotal histopathological characteristic of JCVE.
Figure 1: (A) Cortex with juxtacortical lesion (arrow) and massive cystic changes within the white matter (WM). (B) Multiple infected cortical pyramidal neurons (T-Ag = early viral protein, indicating infection, NeuN = neuronal marker).
4.02
Free Neuropathol 3:20:46
Reduction of oligodendrocyte populations in patients with late-onset multiple sclerosis
Schirin Stephan1, Lidia Stork1, Wolfgang Brück1, Christine Stadelmann-Nessler1, Imke Metz1
1 University Medical Center Göttingen, Institute of Neuropathology, Göttingen, Deutschland
Background: Over 2.8 million people worldwide suffer from multiple sclerosis (MS) - a chronic inflammatory demyelinating disease of the CNS. Typical or normal-onset MS (NOMS) manifests in young adults (20-40 years old). In 3-12% of cases, the disease begins in elderly patients >50 years of age (late-onset MS or LOMS). Such patients more often suffer from primary progressive MS, faster disease progression, and worse recovery from relapses, as well as age-related comorbidities. Animal studies suggest a less efficient remyelination in elderly animals. Marked remyelination, which is observed in 23-50 % of MS lesions in young adult-onset patients, could be slower and less successful in LOMS. A recent study showed that cells expressing BCAS1 represent a subpopulation of actively myelinating oligodendrocytes, and can serve as a reliable marker of ongoing remyelination.
Questions: We addressed the question, if myelin regeneration in LOMS patients is less effective.
Methods: We performed histological analyses of three oligodendrocyte markers that correspond to different oligodendrocyte maturation stages (Olig2 positive cells with a strong nuclear signal for oligodendrocyte precursor cells, BCAS1 for myelinating oligodendrocytes, and NogoA for mature oligodendrocytes) in biopsy specimens from LOMS patients (n=30), and compared them with NOMS patients (n=25).
Results: The number of mature oligodendrocytes was significantly lower in the normal appearing (non-demyelinated) (p=0.02) and perilesional white matter (p<0.0001) in patients with LOMS compared to NOMS. Moreover, mature oligodendrocytes in these regions showed a negative correlation with the age of patients (r=-0.5, p=0.01). There were no differences in the number of oligodendrocyte precursor cells between the two groups. Also, the population of active myelinating BCAS1+ oligodendrocytes was significantly lower in LOMS compared to NOMS patients in the normal-appearing white matter (p=0.03). Again, the number of myelinating cells correlated negatively with the age of patients (r=-0.5, p=0.01). In both groups, the highest number of active myelinating oligodendrocytes was found in early active demyelinating lesions and here at the lesion edge. Although no significant differences in the number of active myelinating cells was found in lesion areas comparing LOMS and NOMS, a trend for higher numbers of myelinating cells was observed in the center of late active lesions in NOMS. In general, older lesions (late active demyelinating and inactive lesions) showed lower numbers of active myelinating cells. LOMS patients had a significantly higher EDSS score (median 3.5, p=0.003) at last follow-up as compared to NOMS patients (median 2.0). A higher EDSS score was associated with a lower number of both mature and oligodendrocyte precursor cells in active MS lesions (r=-0.42, p=0.01 and r=-0.54, p=0.002 respectively).
Conclusions: We observed a significant reduction of active myelinating and mature oligodendrocytes in the periplaque and normal appearing white matter of LOMS patients. This may result in a lower remyelination within lesions. Our results suggest a more efficient remyelination in NOMS compared to LOMS. Importantly, we show that a higher EDSS at last follow-up in LOMS patients negatively correlates with the number of oligodendrocytes within lesions, emphasizing the importance of a loss of oligodendrocytes for clinical disability.
Study supported by Sanofi Genzyme.
4.03
Free Neuropathol 3:20:47
Schwann cell remyelination is a salient feature of spinal NMO with neuroprotective potential
Carolina Thomas1,2, Josephine Sophia Owens1, Parinaz Yavarzadeh1, Anne Winkler1, Christine Stadelmann1
1 Institut für Neuropathologie - Universitätsmedizin Göttingen, Göttingen, Deutschland
2 Max-Planck-Institut für Multidisziplinäre Naturwissenschaften, City Campus - Abteilung Molekulare Neurobiologie, Göttingen, Deutschland
Background: Neuromyelitis optica (NMO) is a severe autoimmune demyelinating disorder characterized by the presence of pathogenic autoantibodies against the water channel aquaporin-4, and clinically by extensive myelitis with optic neuritis. Intriguingly, despite the complete loss of astrocytes with consecutive damage and reduction of oligodendrocytes, a partial remyelination of NMO lesions has been previously described.
Aims: Here we aimed to decipher the cellular components present in remyelinated spinal NMO lesions and to detect potential protective effects of remyelination on axonal integrity in these lesions.
Methods: We performed post-mortem histopathological and immunohistochemical examination of spinal lesions from a cohort of 8 patients.
Results: We demonstrate that Schwann cells (SC) are present and are in part responsible for remyelination of NMO lesions. Furthermore, we show a greater abundance of remyelination in the lesions localized along peripheral nerve entry-exit zones as well as perivascularly. In addition, no SC remyelination was observed in spinal lesions of patients suffering from multiple sclerosis. Finally, axonal density was partially preserved in SC remyelinated areas.
Conclusions: Taken together, our results indicate that SCR is an exclusive feature of NMO that requires a breaching of the glia limitans with potential neuroprotective effects.
5. Muscle / Nerve
5.01
Free Neuropathol 3:20:48
Molecular profiling of skeletal muscle in infantile, juvenile and adult patients with Pompe disease
Alexander Schaiter1, Andreas Roos2,3,4, Andreas Hentschel5, Andreas Hahn6, Marek Bartkuhn7,8, Anne Schänzer1
1 Institute of Neuropathology, Justus Liebig University, Giessen, Deutschland
2 Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, University Duisburg-Essen, Essen, Deutschland
3 Children's Hospital of Eastern Ontario Research Institute; Division of Neurology, Department of Medicine, The Ottawa Hospital; and Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada
4 Department of Neurology, Heimer Institute for Muscle Research, University Hospital Bergmannsheil, Ruhr-University Bochum, Bochum, Deutschland
5 Leibniz-Institute für Analytische Wissenschaften – ISAS – e.V., Dortmund, Deutschland
6 Department of Child Neurology, Justus Liebig University, Giessen, Deutschland
7 Institute of Biomedical Informatics and Systems Medicine Science Unit for Basic and Clinical Medicine, Justus Liebig University, Giessen, Deutschland
8 Institute for Lung Health (ILH) Platform for Genomics and Bioinformatics, Justus Liebig University, Giessen, Deutschland
Introduction: Pompe disease is a lysosomal metabolic disease caused by mutations in the alpha 1,4-glucosidase (GAA). The GAA enzyme defect leads to accumulation of glycogen in striated muscles and a reduced muscle function. Pompe disease is categorized in infantile onset (IOPD) and late onset (LOPD) based on GAA residual activity, genetic profile and clinical manifestation. Furthermore, a juvenile onset is described, which is classified as LOPD. The underlying mechanisms of Pompe disease are still unknown. Molecular signatures of IOPD, LOPD and juvenile patients are rare. The aim of this study is to identify specific differences between the subgroups of Pompe disease using proteomic analysis. The first objective is to find novel protein expressions associated with the subgroups of Pompe disease. The second objective is to compare the proteomic profiles of the subgroups with each other to prove the categorization by a proteomic approach.
Material and Methods: Skeletal muscle biopsies from 35 patients with Pompe disease before start of Enzyme replacement therapy (ERT) (LOPD n=22, IOPD n=11, juvenile n=2) and 21 age matched controls (adult n=11, infantile n=10) were analysed using mass-spectrometry in an LFQ (label-free-quantification) experiment. To identify significantly up or down regulated proteins between the groups, the raw proteomic data was further processed using Maxquant, Perseus and the R-Programming language. Principal Component Analysis (PCA) and Hierarchical Clustering was used to show statistical difference between the sample groups. Further, a pathway analysis of the significantly associated proteins using the KEGG and the gene ontology (GO) library was conducted. To validate associated pathways on another approach a gene set enrichment analysis (GSEA) was calculated. To compare the proteomic profile of LOPD, IOPD and juvenile, the most significant proteins of each group were defined as gene-sets in GSEA.
Results: In the IOPD 13 proteins were up-regulated and 11 downregulated compared to controls. Analyses of juvenile Pompe showed 7 up-regulated, 37 downregulated and in LOPD 19 up-regulated, 47 down-regulated proteins. The samples of LOPD and juvenile shared certain protein expression e.g. FBN1, COL1A2 and COL3A1 were down-regulated in both groups. However, IOPD and LOPD showed divergent expression e.g. HK1 (phosphorylation of various hexoses and involved in innate immunity) was up-regulated in LOPD and down-regulated in IOPD whereas USMG5 (upregulated during skeletal muscle growth) was upregulated only in IOPD. The GSEA further shows a positive correlation between LOPD and juvenile Pompe and a negative correlation between IOPD and LOPD/juvenile Pompe.
Discussion: Mass-spectrometry analysis showed a significant altered regulation in skeletal muscle samples from patients with Pompe disease compared to controls. Interestingly most of the dysregulated proteins varied in IOPD and LOPD subgroup, whereas the expression profiling was similar in the juvenile patients compared to LOPD. These data underline the heterogeneity in Pompe disease and may indicate a divergent underlying muscle pathology in infantile patients with more immature skeletal muscle fibers compared to juvenile and adult patients.
5.02
Free Neuropathol 3:20:50
Expression of immune regulating proteins in skeletal muscle of different idiopathic inflammatory myopathies (IIM) subtypes
Anna Nishimura1, Rebecca Hasseli2,3, Heidrun H. Krämer4, Angela Roth1, Eva Neuen-Jacob5, Tobias Ruck6, Anne Schänzer1
1 Institute of Neuropathology, Justus-Liebig-University, Giessen, Deutschland
2 Department of Rheumatology and Clinical Immunology, Campus Kerckhoff, Justus-Liebig-University, Giessen, Deutschland
3 Department of Internal Medicine II, Justus-Liebig-University, Giessen, Deutschland
4 Department of Neurology, Justus-Liebig-University, Giessen, Deutschland
5 Institute of Neuropathology, Heinrich-Heine-University, Düsseldorf, Deutschland
6 Department of Neurology, Medical Faculty, Heinrich-Heine-University, Düsseldorf, Deutschland
Background: Idiopathic inflammatory myopathies (IIMs) are autoimmune diseases classified as polymyositis (PM), dermatomyositis (DM), immune-mediated necrotizing myopathy (IMNM), anti-synthetase syndrome (ASyS) and sporadic inclusion body myositis (sIBM). Adaptive and innate immune responses play a role in the pathogenesis of IIM and immunomodulatory treatment is the recommended therapy. The individual regulatory mechanisms in IIM subtypes might differ and a better understanding of the pathophysiology would improve the individual therapeutic approaches. With whole muscle section morphometry, we want to analyze the immune regulating proteins in certain subtypes of IIM.
Methods: Muscle biopsies from 24 adult patients with IIM (average age at biopsy 54.6 years; 65.2 % female; DM n=5; IMNM n=5; ASyS n=7; sIBM n=7), neurogenic atrophy (NA; n=4) and controls (HC; n=6) were included in the study. All biopsies were re-evaluated according to the common classification. The degree of pathology severity was estimated using a semiquantitative pathology score (p-score from 0 to 10). Double immunofluorescence staining was performed with antibodies against MHC-1 (major histocompatibility complex 1), MHC-2, ICAM-1 (intercellular adhesion molecule 1) and VCAM-1 (vascular cell adhesion molecule 1) and antibodies against spectrin or desmin. The sections were subsequently digitalized using a Zeiss Axio Scan.Z1 slide scanner. The coexpression was analyzed on entire sections using ImageJ software and quantified using the Manders' coefficient (M).
Results: MHC-1 expression was significantly upregulated on muscle fibers of patients with ASyS (M=0.559), DM (M=0.609), sIBM (M=0.557) and NA (M=0.208) compared to HC (M=0.009). For IMNM (M=0.079), the expression was significantly lower compared to ASyS and DM. Significant upregulations of MHC-2 were found for ASyS (M=0.263), DM (M=0.504), sIBM (M=0.336) and NA (M=0.416) compared to HC (M=0.036), as well as a significant upregulation for DM compared to IMNM (M=0.055). Only ASyS samples showed a significant upregulation of ICAM-1 compared to HC (M=0.060), whereas in general, the expression varied strongly (ASyS M=0.263±0.182; DM M=0.270±0.256; sIBM M=0.337±0.249; IMNM M=0.335±0.295) with no significant difference between the IIM subtypes. VCAM-1 expression showed no significant difference between the IIM subtypes. However, in comparison to HC (M=0.036) we found VCAM-1 significantly upregulated in ASyS (M=0.200), DM (M=0.377) and sIBM (M=0.371). Correlating our results with the p-score, a positive correlation (r=0.770) for ICAM-1 expression in sIBM was found.
Conclusion: We analyzed protein expression levels on muscle fibers in whole section analysis, which provides accurate data on larger sections. Different expression patterns of MHC-1/2, ICAM-1 and VCAM-1 were found in IIM subtypes with lower expressions in IMNM for MHC-1/2 and VCAM-1. Interestingly, in sIBM ICAM-1 expression correlated with the pathology score. These insights might help to improve morphological diagnosis in IIM subtypes and identify individual immune response patterns, which may improve the accuracy of future diagnoses.
5.03
Free Neuropathol 3:20:52
Long Term Safety and Efficacy Outcomes for X-Linked Myotubular Myopathy (XLMTM) with Gene Replacement Therapy, Resamirigene Bilparvovec (ASPIRO): Preliminary Results from Cohort 1 in ASPIRO, a Phase 1/2/3 Study
Astrid Blaschek1, Perry Shieh2, Nancy Kuntz3, James Dowling4, Carsten Bonnemann5, A. Reghan Foley5, Dimah Saade6, Andreea Seferian7, Laurent Servais8, Neema Lakshman9, Cong Han10, Suyash Prasad11, Salvador Rico11, Westin Miller9
1 Klinikum der Universität München, München, Deutschland
2 University of California, Los Angeles, United States
3 Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, United States
4 The Hospital for Sick Children, Toronto, Canada
5 National Institutes of Health, Bethesda, United States
6 University Of Iowa Hospitals and Clinics - Medicine Specialty Clinics, Iowa City, United States
7 Institut de Myologie, Paris, France
8 MDUK Oxford Neuromuscular Centre, Oxford, United Kingdom
9 Astellas Gene Therapies, California , United States
10 Astellas Pharma Global Development, Northbrook, United States
11 Formerly Astellas Gene Therapies, California, United States
Background: XLMTM is a rare, life-threatening congenital myopathy caused by mutations in the MTM1 gene. There is no approved treatment for XLMTM.
Objectives: ASPIRO (NCT03199469), a Phase 1/2/3 randomized, open-label study is investigating the safety and efficacy of AT132 (resamirigene bilparvovec), a single-dose gene replacement therapy for ventilatory-dependent XLMTM.
Question: What is the primary safety and efficacy data from ASPRIO?
Methods: Participants were young boys with genetically confirmed XLMTM. The primary efficacy outcome was the change in hours of daily ventilator support from baseline through Week 48. The key secondary efficacy outcome was percentage of participants who achieve functionally independent sitting by Week 48. We report long-term safety and key efficacy outcomes (up to 42 months [m]) for the first 6 participants dosed in ASPIRO, all receiving the lower-dose of 1.3 x 1014 vg/kg and compared with 15 untreated controls (including 12 participants from INCEPTUS), as of 29JAN2021.
Results: The mean age at dosing was 20·4m (range: 9·5-49·7m) and 19·6m (5·9-39·3m) at enrollment among dosed participants and controls, respectively. Major developmental milestones achieved by all dosed participants over time is shown in Figure 1. All dosed participants were ventilator dependent at first assessment; 5 (83.33%) requiring transtracheal invasive ventilation >22 hours/day and 1 (16.67%) used non-invasive ventilation of 12 hours/day. All dosed participants achieved ventilator independence, 5 remain so (mean durability 25.6m; range 18.3-36.6m) of which 4 have been decannulated. No control participants became ventilator independent or were decannulated.
At baseline, 1/6 dosed participant was able to sit independently without support for 30 seconds; 5/6 participants did not have full head control and were unable to sit independently. Major motor milestones were achieved in all dosed participants (Figure 1); 5/6 remain independently ambulatory without assistive device (achieved mean [SD] time 21.92 [5.57]m); 4 of whom have achieved the ability to ascend stairs. 5/15 (33.3%) control participants achieved independent sitting; none achieved higher milestones.
Among 6 dosed in cohort 1, 4 (67%) participants experienced treatment-emergent severe adverse events; infections in 4 (67%) and respiratory/thoracic/mediastinal disorders in 1(17%). All dosed participants currently have stable liver function test values. As of January 2021, three deaths in the higher-dose cohort occurred following severe decompensated liver disease, and three deaths in the control cohort (aspiration pneumonia; cardiopulmonary failure; hepatic hemorrhage with peliosis) were observed. As of September 2021, a newly dosed participant in the lower-dose cohort experienced severe liver function test abnormalities and has died.
Conclusions: A rapid improvement in respiratory and motor outcomes was observed among 6 XLMTM participants dosed with AT132 at 1.3 x 1014 vg/kg vs control participants; these improvements have been maintained and expanded upon over time, indicating improved strength, function, and quality of life for these dosed participants. These substantial improvements must be weighed against the occurrences of fatal serious adverse events, for which the ASPIRO program is on clinical hold while relevant clinical information is being gathered and reviewed.
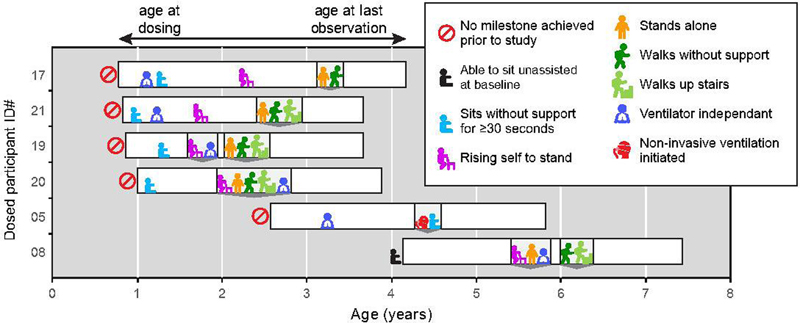
Figure 1: Developmental motor and respiratory milestones achieved in XLMTM patients dosed at AT132 at 1.3 x 1014 vg/kg.
5.04
Free Neuropathol 3:20:54
Lymphotoxin-driven chronic mucle inflammation interdepends with impaired autophagy, self-perpetuates and models inclusion body myositis in mice
Juliane Bremer1, Judith Bauer2, Jana Zschüntzsch3, Thomas Blank4, Kamil Zajt1, Laura Anna Fischer3, Anna Sensmeyer3, Lara Wiechers3, Josef Reichelt3, Kai Hofmann5, Monika Wolf6, Corinna Leuchtenberger5, Priyanka Tripathi1, Claudia Einer7, Hans Zischka7, Adriano Aguzzi6, Regina Reimann6, Veronika Kana6,8, Elisabeth Rushing6, Marco Prinz4, David Liebetanz3, Francesca Odoardi3, Joachim Weis1, Jens Schmidt3,9, Mathias Heikenwälder5
1 Uniklinik RWTH Aachen, Institut für Neuropathologie, Aachen, Deutschland
2 Technische Universität München, Institut für Toxikologie und Umwelthygiene, München, Deutschland
3 Universitätsmedizin Göttingen, Klinik für Neurologie, Göttingen, Deutschland
4 Universitätsklinikum Freiburg, Institut für Neuropathologie, Freiburg, Deutschland
5 DKFZ Heidelberg, Heidelberg, Deutschland
6 Universitätsspital Zürich, Institut für Neuropathologie, Zürich, Switzerland
7 Helmholtz München, Institut für Molekulare Toxikologie und Pharmakologie , München, Deutschland
8 Universitätsspital Zürich, Klinik für Neurologie, Zürich, Switzerland
9 Universitätsklinik der Medizinischen Hochschule Brandenburg, Abteilung Neurologie und Schmerztherapie, Rüdersdorf, Deutschland
Background, goals and research question: Inclusion body myositis (IBM) is a progressive muscular disorder characterized by muscle inflammation and degeneration including protein aggregates and altered autophagic activity. The combination of inflammatory and degenerative features has led to the assumption that this interrelationship may serve as a major driver of the disease pathology. In view of the lack of effective treatment there is an urgent need for useful model systems that reflect the presumed pathomechanisms. A reliable animal model for chronic inflammatory and degenerative features as in IBM has not been available.
Methods/Results: Here, we established mouse models with lymphotoxin expression-driven chronic inflammation of skeletal muscle and with impaired autophagy due to muscle cell specific ATG5-knockout. Both conditions alone induced weakness and muscular atrophy. Gene expression analysis showed that, while chronic inflammation alone drives endoplasmic reticulum stress and alters autophagy/ proteostasis, autophagy disruption alone induces a pro-inflammatory state. This suggests that both, inflammation and autophagy disruption are interdependent in IBM pathogenesis. Only when we genetically combined transgene-driven inflammation with autophagy impairment in mice, skeletal muscle fibers displayed characteristic molecular and neuropathological features of IBM, including protein aggregates with typical ultrastructural morphology. Given that human IBM is refractory to established drugs, we aimed to mirror recent treatment failures and identify the underlying mechanisms by subjecting four months-old myositis mice to corticosteroids, anti-CD52 or anti-Thy1.2 antibodies to deplete lymphocytes, or by blocking lymphotoxin beta-receptor signaling. None of these treatments was able to significantly improve muscular performance of the mice or expression profiles of molecular indicators of muscle pathology. This suggests that, once established, IBM-like pathology cannot be reverted or prevented from progression, but is a self-perpetuating condition.
Conclusions: In summary, the data provide unique evidence that inflammation and autophagy disruption are intertwined in IBM-like muscular pathology. It can be expected that this novel mouse model will substantially further our effort to identify better treatment modalities for IBM in the future.
5.05
Free Neuropathol 3:20:56
Novel form of congenital myopathy caused by bi-allelic mutations in uncoordinated mutant number-45 myosin chaperone B
Sebahattin Cirak1
1 Uniklinik Ulm, Klinik für Kinder-und Jugendmedizin, Sozialpädiatrisches Zentrum und Pädiatrische Neurologie mit Stoffwechsel, Ulm, Deutschland
Background: Congenital myopathies (CM) form a genetically heterogeneous group of disorders, only 60% can be genetically solved.
Aims: Discovery of novel genetically defined myopathies.
Question: Deciphering the genomic landscape of congenital myopathies.
Methods: We recruited an 11-year old male of consanguineous parents, presenting with proximal weakness, Gower's sign, without cardiomyopathy with a stable disease course. We performed exome sequencing and data analysis was performed with our in-house software Varbank2 according to an autosomal recessive inheritance. We investigated the effect of the missense mutation by complementation assay on the zebrafish steif mutant, an unc-45b loss-of-function model.
Results: We have discovered and published a novel genetically defined form of CM due to a novel homozygous missense mutation in UNC45B (NM_173167.2: c.2261G>A, p.Arg754Gln) also co-segregating in the family with three healthy siblings (Dafsari et al., 2019). In our patient's muscle biopsy, core-like structures were detected mainly in the center of muscle fibers in NADH histochemistry. Electron microscopy showed numerous focal core-like alterations of myofibrillar architecture with Z-bands streaming.
Conclusions: Three isoforms of UNC45B are highly expressed in skeletal muscle, only one also in cardiac muscle. Due to its high evolutionary conservation throughout species, a loss of UNC45 results in different pathological conditions in various species: a knockdown of unc-45 resulted in dilated cardiomyopathy and a reduced muscle contractility in D. melanogaster. Similarly, in unc-45b knockdown zebrafish and also in steif mutants, disrupted myofibrillogenesis associated cardiac dysfunction and paralysis was observed. Injection of mutant unc-45b mRNA did not rescue the steif mutant in contrast to wt mRNA confirming the pathogenicity of the missense mutation.
6. Free Topics
6.01
Free Neuropathol 3:20:57
Deep genotype-phenotype analysis of Focal Cortical Dysplasia type 2 differentiates between a GATOR-positive autophagy altered subtype 2a and MTOR-positive migration deficit subtype 2b
Jonas Honke1, Lucas Hoffmann1, Roland Coras1, Javier A. López-Rivera2,3,4, Costin Leu3,4,5,6, Dennis Lal3,4,5,6, Peter Nürnberg6, Tom Pieper7, Till Hartlieb7, Manfred Kudernatsch7, Christian G. Bien8, Friedrich Woermann8, Thomas Cloppenborg8, Hajo Hamer9, Sebastian Brandner10, Karl Rössler11, Arnd Dörfler12, Stéphanie Baulac13, Sara Baldassari13, Ingmar Blümcke1,4
1 Department of Neuropathology, Universitätsklinikum Erlangen, FAU Erlangen-Nürnberg, and partner of the European Reference Network (ERN) EpiCARE , Erlangen, Deutschland
1 Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Case Western Reserve University, Cleveland, United States
1 Genomic Medicine Institute, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195, United States
1 Charles Shor Epilepsy Center, Neurological Institute, Cleveland Clinic, Cleveland, United States
1 Stanley Center for Psychiatric Research, Broad Institute of Harvard and M.I.T, Cambridge, MA 02142, United States
1 Cologne Center for Genomics (CCG), Medical Faculty of the University of Cologne, University Hospital of Cologne, 50931 Cologne, Germany
1 Center for Pediatric Neurology, Neurorehabilitation, and Epileptology, Schoen-Clinic, Vogtareuth, Germany
1 Department of Epileptology (Krankenhaus Mara), Medical School, Bielefeld University, Bielefeld, Germany
1 Epilepsy Center, Universitätsklinikum Erlangen, FAU Erlangen-Nürnberg, and EpiCARE partner, Erlangen, Germany
1 Department of Neurosurgery, Universitätsklinikum Erlangen, FAU Erlangen-Nürnberg, Erlangen, Germany
1 Department of Neurosurgery, Medical University of Vienna, Vienna General Hospital, Vienna, Austria
1 Department of Neuroradiology, Universitätsklinikum Erlangen, FAU Erlangen-Nürnberg, Erlangen, Germany
1 Sorbonne Université, Institut du Cerveau - Paris Brain Institute - ICM, Inserm, CNRS, APHP, Hôpital de la Pitié Salpêtrière, Paris, France
Background: Focal Cortical Dysplasia Type 2 (FCD2) is the single most common cause of drug-resistant focal epilepsy in children. Despite continuous progress in diagnostic methods, however, the recognition and treatment of FCD2 subtypes remain a challenging issue in clinical practice. Herein, we performed a deep genotype-phenotype analysis to further elucidate the clinical-pathological and genetic presentation of FCD2.
Methods: Patients with focal epilepsy submitted to neurosurgical treatment, histopathological confirmed diagnosis of FCD ILAE Type 2 and positive genetic testing were retrieved from the European Epilepsy Brain Bank. Clinical data were available from the contributing epilepsy centers. Deep whole-exome sequencing with a coverage of >350X or mTOR gene panel sequencing with a coverage of >1000X were obtained from fresh frozen tissue samples. Histopathological analyses were performed from formalin-fixed, paraffin embedded tissue samples using HE and immunohistochemistry for NF-SMI32, NeuN, pS6, p62, and Vimentin. All slides were digitalised and further analysed with QuPath v.0.3.0.
Results: Seventeen patients were identified by carrying pathogenetic variants in genes directly associated with the mTOR pathway, i.e., loss-of-function in the GATOR1 complex (DEPDC5, n=7 and NPRL3, n=3), or gain-of-function in the mTORC1 signalling pathway [MTOR, n=7]. All patients were seizure-free after surgery with the exception of four patients carrying a DEPDC5 mutation. Histopathological analysis revealed a FCD2A subtype in all cases with GATOR1 alteration, i.e., no balloon cells. In contrast, the FCD2B subtype was predominant in cases with gene variants affecting the mTORC1 signalling complex. Specimens carrying MTOR variants also had significantly larger dysmorphic neurons than GATOR1 variants [p=0.005]. In addition, five cases defined by GATOR1 variants showed a unique and predominantly vacuolizing phenotype (Figure 1C). All cases with GATOR1 alterations were located in the frontal lobe and the majority was confined to the cortical ribbon not affecting the white matter (Figure 1A). This pattern was reflected by subtle or negative MRI findings in 9/10 patients with GATOR1 variants.
Discussion/Conclusion: We describe a yet unrecognized genotype-phenotype correlation of GATOR1 variants with FCD2A in the frontal lobe. These lesions were histopathological further characterized by abnormally vacuolizing cells suggestive of an autophagy altered phenotype. From recently published evidence, we also hypothesize that this subtype will carry a second hit brain somatic variant in the DEPDC5 gene, which was, however, difficult to identify with techniques applied in our current study. In contrast, patients with FCD2B and brain somatic MTOR variants showed larger lesions on MRI including the white matter, suggesting compromised neural migration (Figure 1B). These data may help to better understand difficult-to-diagnose and treat FCD subtypes, i.e., ILAE Type 2A and 2B.
Figure 1: Genotype-phenotype analysis of FCD ILAE Type 2
A: 42-year-old male patient with frontal lobe epilepsy since age 5 years, histopathological confirmed FCD2A and a pathogenic DEPDC5 mosaicism. The arrow points to the sharp border between the cortical FCD and the normal appearing white matter (WM). NCx - normal 6-layered neocortex. Neurofilament SMI32 immunohistochemistry. Scale bar = 2.5mm (applies also to B). Higher magnification in C reveals dysmorphic neurons with a vacuolizing predominant phenotype (red arrow) suggesting an autophagy altered phenotype. Scale bar = 100µm (applies also to D). B: 19-year-old male patient with frontal lobe epilepsy since age 9 years, histopathological confirmed FCD2B at a bottom-of-sulcus (BoS; higher magnification in D) and a pathogenic MTOR mosaicism. Dysmorphic neurons and balloon cells (latter not shown) were aggregating in the neocortex and white matter (arrow) suggesting a migration deficient phenotype.
6.02
Free Neuropathol 3:20:60
Age-dependent increase of perineuronal nets in the human hippocampus of patients with and without temporal lobe epilepsy
Annika Lehner1, Lucas Hoffmann1, Friedrich Paulsen2, Hajo Hamer3, Katrin Walther3, Karl Rössler4, Sebastian Brandner5, Ingmar Blümcke1
1 Universitätsklinikum Erlangen and FAU Erlangen-Nürnberg, Department of Neuropathology, Erlangen, Deutschland
2 FAU Erlangen-Nürnberg, Institute of Functional and Clinical Anatomy, Erlangen, Deutschland
3 Universitätsklinikum Erlangen and FAU Erlangen-Nürnberg, Epilepsy Center, Neurological Institute, Erlangen, Deutschland
4 Medical University of Vienna, Vienna General Hospital, Department of Neurosurgery, Vienna, Austria
5 Universitätsklinikum Erlangen and FAU Erlangen-Nürnberg, Department of Neurosurgery, Erlangen, Deutschland
Background: Perineuronal nets (PNN) are a specialized extracellular matrix surrounding Parvalbumin-positive GABAergic interneurons of the central nervous system, contain mainly aggrecan and lecticans, and play a role in the regulation of synaptic plasticity, brain maturation and cognitive impairment. In the human neocortex, PNN appear in their full expression at an age of eight, and alterations of PNN have been found in neurological disorders including Alzheimer´s disease, schizophrenia or epilepsy. However, their role in functional maintenance of the CNS as well as disease pathogenesis remains to be elucidated.
Aim: Our aim was to histopathologically assess PNN in the hippocampus of patients with temporal lobe epilepsy (TLE) compared to age-matched post-mortem control subjects.
Methods: Formalin-fixed and paraffin-embedded neurosurgical tissue specimens of the human hippocampus were retrieved from the European Epilepsy Brain Bank. Twenty-nine patients had histopathologically confirmed hippocampal sclerosis (HS), and 11 patients suffered from TLE with no HS. Neuropsychology scores for cognitive tasks were retrieved from the files of the Erlangen Epilepsy center. Hippocampus samples of 27 postmortem brains served as control (age range 3-84). PNN were visualized using antibodies directed against aggrecan (Figure 1). PNN were manually recorded in the dentate gyrus, CA1, CA2, CA3, CA4 and subiculum. PNN density per mm² was then compared between different HS subtypes according to the ILAE classification scheme and controls. Selected cases were double immunofluorescence labeled with antibodies against parvalbumin and aggrecan (Figure 1) and further analyzed on fully digitalized scans (Hamamtsu NanoZoomer S60) with QuPath 0.3.0.
Results: The density of PNN increased with age in both, human controls and TLE patients (Figure 2). However, the density of PNN was significantly higher in all TLE patients compared to age-matched controls with a non-significant increase in patients with hippocampal sclerosis compared to TLE patients without HS. These alterations were also region-specific and most obvious in the subiculum, CA1 and dentate gyrus (Figure 3). There was no significant correlation with cognitive impairment in our cohort of TLE patients.
Discussion: We quantitatively described the normal distribution of PNN in the human hippocampal formation. We investigated changes with age in the number of perineuronal nets in older patients and found new evidence that epilepsy alters perineuronal nets.
Figure 1: Triple immunofluorescence-staining at 40x magnification: a parvalbumin-positive Neuron surrounded by Aggrecan (left side). Immunohistochemical staining of a PNN in the Stratum plexiforme of the dentate gyrus (150 µm away from Stratum granulosum), Screenshot from QuPath, 40x magnification (right side).
Figure 2: correlation between the age and the average number of PNN per mm² in patients with and without epilepsy
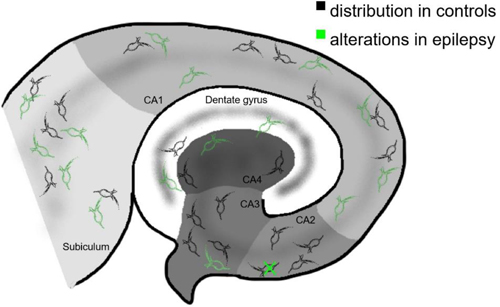
Figure 3: the normal distribution of PNN and alterations in epilepsy
6.03
Free Neuropathol 3:20:63
Vakuolisierung der Dura als nicht-lymphassoziierte Veränderung
Ralf Schober1, Christian Eisenlöffel1, Max Holzer2
1 Klinikum St. Georg, Inst. f. Pathologie u. Tumordiagnostik, Leipzig, Deutschlandy
2 Paul-Flechsig-Institut für Hirnforschung, Univ. Leipzig, Leipzig, Deutschland
Die meningealen Lymphgefäße stellen eine Zwischenstation beim Abtransport des Liquors vom glymphatischen System des Gehirns zu den zervikalen Lymphknoten dar (Louveau J ea 2015, Aspelund A ea 2015). Sie gelten als wichtiges Immun-Portal für das zentrale Nervensystem (Tavares GA u. Louveau A 2021), und eine Verlegung des glymphatischen Systems wird pathogenetisch mit der Demenz und insbesondere der Alzheimer'schen Erkrankung in Verbindung gebracht (Nedergaard M u. Goldman SA 2020). Histologische Darstellungen der überwiegend Sinus-nah in der Dura gelegenen Lymphkanäle beziehen sich bislang hauptsächlich auf die Ratte, detailliertere Untersuchungen beim Menschen und insbesondere bei verschiedenen Erkrankungen stehen noch aus.
Wir haben in 35 Sektionsfällen die Dura untersucht, meist im Bereich des Sinus sagittalis superior, teilweise zusätzlich des Sinus transversus. Das Alter der 14 weiblichen und 21 männlichen Patienten und Patientinnen betrug 25-91 Jahre, im Durchschnitt 69 Jahre. In 8 Fällen war ein Morbus Alzheimer diagnostiziert worden, in 9 Fällen Alzheimer-assoziierte Veränderungen, in je einem Fall PART und Morbus Parkinson. 6 Fälle hatten meningitische Veränderungen, 2 Fälle eine Meningeosis carcinomatosa, 2 Fälle eine AV-Fistel, 3 Fälle intra-/subdurale Blutungen, und 3 Fälle eine Sinusthrombose. Im histologischen Bild vorherrschend und in allen außer 3 Fällen nachweisbar waren jedoch nicht die Lymphgefäße, sondern Gruppen kleiner intraduraler Vakuolen, 30-300 µ im Durchmesser, meist glatt begrenzt und ohne Zellbelag. Bei immunhistochemischen Untersuchungen in ausgewählten Fällen zeigten sie keine positive Reaktion auf Podoplanin, CD31, CD34, S100, Aβ und APP. Dagegen ließen sich Lymphgefäße deutlich anhand ihrer positiven Reaktion auf Podoplanin identifizieren, sie erschienen durchweg streifenförmig-schmal und regelrecht strukturiert.
Die Anzahl der Vakuolen-Gruppen und ihre Dichte in der Dura wurde semiquantitativ in 6 Stufen von nicht vorhanden bis stark eingeteilt, und diese Stufen wurden jeweils den Falldaten gegenübergestellt. Eine Korrelation mit bestimmten Erkrankungen ergab sich nicht, lediglich eine partielle Korrelation mit dem Alter. Deutlich vermehrt waren Vakuolen bei gleichzeitigem Nachweis einer größeren Anzahl fibrosierter Pacchionischer Granulationen vorhanden.
Zusammenfassend stellt eine Vakuolisierung der Dura im Sinusdach-Bereich eine relativ häufige Veränderung dar, nicht im Zusammenhang mit dem Lymphsystem stehend sowie offenbar nicht Krankheits-korreliert und am ehesten degenerativer Natur. Die Befunde bestätigen und ergänzen kurze Angaben der älteren Literatur, wo zunächst von Grüppchen großblasiger Zellen gesprochen wird (Schaffer J 1933), dann aber von Altersveränderungen mit Hohlraumbildungen durch Aufquellung und Zerfall von Bindegewebsfasern (Schaltenbrand G u. Dorn E 1955).
6.04
Free Neuropathol 3:20:64
MOGHE with or without SLC35A2 brain somatic mutations reveal a common phenotype of oligodendroglial regeneration and remyelination
Simon Geffers1, Lucas Hoffmann1, Roland Coras1, Katja Kobow1, Javier Lopez-Riviera2,3,4, Costin Leu3,4,5,6, Dennis Lal3,4,5,6, Peter Nürnberg6, Stephanie Bulac7, Till Hartlieb8,9, Tom Pieper8, Manfred Kudernatsch8,10, Christian Bien11, Hajo Hamer12, Karl Rössler13,14, Sebastian Brandtner14, Christine Stadelmann15, Ingmar Blümcke1
1 Department of Neuropathology, University Hospital Erlangen, Erlangen, Deutschland
2 Department of Molecular Medicine, Cleveland Clinic Lerner College of Medicine, Cleveland, United States
3 Genomic Medicine Institute, Lerner Research Center, Cleveland Clinic, Cleveland, United States
4 Charles Shor Epilepsy Center, Neurological Institute, Cleveland Clinic, Cleveland, United States
5 Stanley Center for Psychiatric Research, Broad Institute of Harvard and M.I.T, Cambridge, United States
6 Cologne Center for Genomics (CCG), Medical Faculty of the University of Cologne, University Hospital of Cologne, Cologne, Deutschland
7 Sorbonne Université, Institut du Cerveau , Paris Brain Institute, Paris, France
8 Center for Pediatric Neurology, Neurorehabilitation and Epileptology, Schoen Klinik Vogtareuth, Vogtareuth, Deutschland
9 Research Institute Rehabilitation, Transition, Palliation, PMU Salzburg, Salzburg, Austria
10 Center for Neurosurgery, Epilepsy Surgery, Spine Surgery and Scoliosis, Schoen Klinik Vogtareuth, Vogtareuth, Deutschland
11 Department of Epileptology (Krankenhaus Mara), Bielefeld University, Bielefeld, Deutschland
12 Epilepsy Center, University Hospital Erlangen, FAU Erlangen Nürnberg, Erlangen, Deutschland
13 Department of Neurosurgery, University Hospital Erlangen, FAU Erlangen Nürnberg, Erlangen, Deutschland
14 Department of Neurosurgery, Medical University of Vienna, Vienna General Hospital, Vienna, Austria
15 Institute of Neuropathology, University Medical Center Göttingen, Göttingen, Deutschland
Mild malformation of cortical development with oligodendroglial hyperplasia in epilepsy (MOGHE) is a recently identified disease entity associated with early onset frontal lobe epilepsy in the vast majority of reported cases. Brain somatic mutations in the UDP-galactose transporter SLC35A2 gene were identified in about 50% of published MOGHE cases as underlying pathogenetic variant. Herein, we performed a phenotype-genotype analysis of MOGHE with SLC35A2 mutations compared to MOGHE without SLC25A2 mutations in order to assess a potentially different phenotype-genotype relationship at the microscopy and clinical level. This may also help us to further untangle the yet unknown aetiological spectrum of the MOGHE disease.
We retrieved 39 surgical brain specimens histopathologically characterized as MOGHE from the archives of the European Epilepsy Brain Bank, 19 with and 20 without brain somatic SLC35A2 variants and compared them with 20 age-matched controls, i.e., FCD 2A (n=10) and non-epileptic post-mortem specimens of the frontal neocortex (n=10). Semi-quantitative analysis using a panel of immunohistochemical markers addressing oligodendroglial lineage (OLIG2), maturation (Breast cancer amplifying sequence 1; BCAS1), proliferation (Ki67), and myelination (CNPase, Nissl-LFB) were performed.
The clinical presentation was not significantly different between both study cohorts (MOGHE with or without SLC35A2 mutations), e.g., median seizure onset was at 1.2 and 1.8 years, respectively. Patients were operated at 6.3 and 7.5 years, respectively, and 89% of lesions were localized in the frontal lobe. However, patients with SLC35A2 mutated MOGHE presented with more infantile spasms (61% vs. 25%). Patchy areas of increased oligodendroglial cellularity and decreased myelination were matching each other and were present in both groups (Figure 1). In addition, regenerative oligodendrocytes immunoreactive for BCAS1 were visible in both MOGHE groups and both groups had higher numbers of BCAS-positive oligodendrocytes compared to our age-matched control cohort (Figure 2). Interestingly, MOGHE patients operated at an older age (>17 years; n=5) showed lower BCAS and oligodendroglial cell numbers which were similar to those observed in our post-mortem control cohort. Myelination deficits were also less visible in MOGHE patients operated at an older age (Figure 3).
In conclusion, our results revealed a similar genotype-phenotype correlation of MOGHE w/o SLC35A2 brain somatic mutations. These results further suggest a similar oligodendroglial regeneration and remyelination capacity of early-onset MOGHE subtypes, and that the same UDP-galactose transport pathway is likely to play a role in MOGHE with SLC35A2 wildtype urging for a more systematic approach deciphering the pathogenic aetiology of the MOGHE disease spectrum.
Figure 1: Patchy regions with increased oligodendroglial cell density (heatmap analysis in A, B, E) revealed a matching overlay with patchy areas of hypomyelination (F). MOGHE with SLC35A2 mutations in A, C-F; B – MOGHE without SLC35A2 mutation.
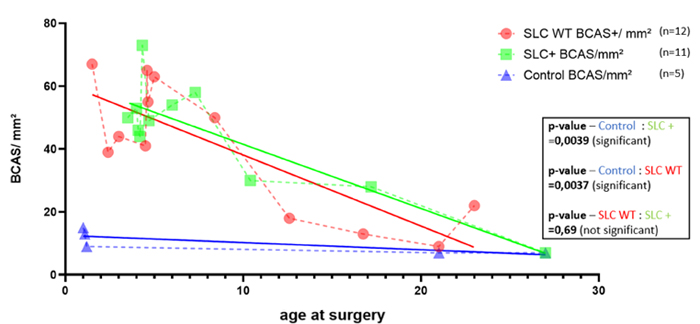
Figure 2: All MOGHE samples revealed a higher number of BCAS1-immunoreactive oligodendrocytes at early ages, which normalize with further maturation.
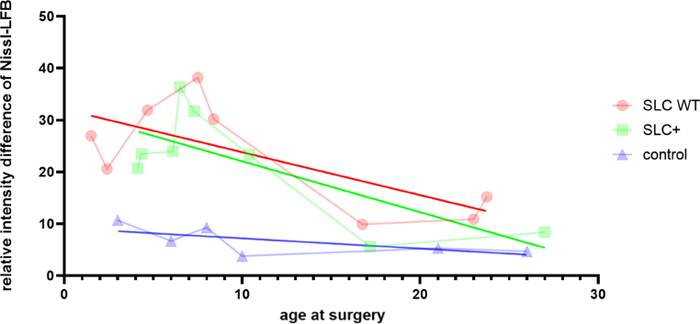
Figure 3: All MOGHE samples revealed a higher degree of hypomyelination (Nissl-Luxol-Fast-Blue, LFB) at early ages, which normalize with further maturation.
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
|
