|
|
|
Free Neuropathology 3:21 (2022) |
|
Case Report |
|
Malignant melanotic nerve sheath tumor with PRKAR1A, KMT2C and GNAQ mutations |
|
Merryl Terry1, Kristina Wakeman1, Brian J. Williams2,4, Donald M. Miller3,4, Müge Sak5, Zied Abdullaev6, Marwil C. Pacheco1, Kenneth Aldape6, Norman L. Lehman1,4,5 |
|
1 Department of Pathology and Laboratory Medicine, University of Louisville, Louisville, KY, USA |
|
Corresponding author: |
|
Submitted: 03 April 2022 Accepted: 12 August 2022 Copyedited by: Jeffrey Nirschl Published: 26 August 2022 |
|
Keywords: Malignant melanotic nerve sheath tumor, MMNST, Psammomatous melanotic schwannoma, Nerve sheath tumor, Pigmented epithelioid melanocytoma, PRKAR1A, KMT2C, GNAQ |
|
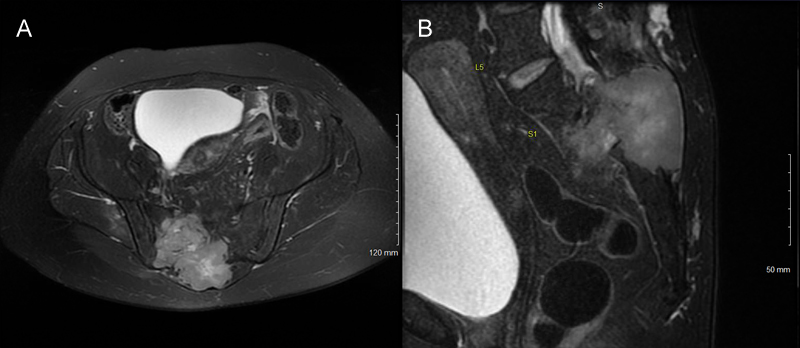
Abstract Malignant melanotic nerve sheath tumor (MMNST) is a rare and potentially aggressive lesion defined in the 2021 WHO Classification of Tumors of the Central Nervous System. MMNST demonstrate overlapping histologic and clinical features of schwannoma and melanoma. MMNST often harbor PRKAR1A mutations, especially within the Carney Complex. We present a case of aggressive MMNST of the sacral region in a 48-year-old woman. The tumor contained PRKAR1A frame shift pR352Hfs*89, KMT2C splice site c.7443-1G>T and GNAQ p.R183L missense mutations, as well as BRAF and MYC gains. Genomic DNA methylation analysis using the Illumina 850K EpicBead chip revealed that the lesion did not match an established methylation class; however, uniform manifold approximation and projection (UMAP) placed the tumor very near schwannomas. The tumor expressed PD-L1, and the patient was treated with radiation and immune checkpoint inhibitors following en bloc resection. Although she had symptomatic improvement, she suffered early disease progression with local recurrence, and distant metastases, and died 18 months after resection. It has been suggested that the presence of GNAQ mutations can differentiate leptomeningeal melanocytic neoplasms and uveal melanoma from MMNST. This case and others demonstrate that GNAQ mutations may exist in malignant nerve sheath tumors; that GNAQ and PRKAR1A mutations are not always mutually exclusive and that neither can be used to differentiate MMNST or MPNST from all melanocytic lesions. Introduction Malignant melanotic nerve sheath tumors (MMNSTs) are rare neoplasms formerly known as melanotic schwannoma [1] and sometimes melanotic malignant peripheral nerve sheath tumor (MPNST). They are renamed in the 2021 WHO CNS tumor classification [2] to better reflect their potentially aggressive nature and align with the 2020 WHO soft tissue nomenclature [3]. MMNST often carry an inactivating mutation in the protein kinase A inhibitory subunit gene protein kinase cAMP-dependent type I regulatory subunit alpha (PRKAR1A) on chromosome 17p22-24, which acts as a tumor suppressor [3, 4]. MMNST occur in some patients with Carney Complex, a rare syndrome associated with PRKAR1A mutation, cardiac and skin myxomas, endocrine and gonadal neoplasms, variable endocrine manifestations such as Cushing syndrome or acromegaly, increased lentigines, and a type of blue nevus known as pigmented epithelioid melanocytoma [5, 6]. We present a case of an aggressive sacral MMNST in a woman which demonstrated PRKAR1A, KMT2C, and GNAQ mutations, BRAF and MYC copy number gains, and PD-L1 overexpression. We also compare histologic, genetic, and clinical features of conventional schwannomas, MMNST, MPNST, and melanoma. Case Report A 48-year-old female developed severe lower back and right buttock pain with right thigh numbness over a period of one year. She also reported difficulty urinating and defecating. Her past medical history included endometriosis, hypothyroidism, and multinodular goiter. On examination, she had saddle anesthesia and urinary retention. Computed tomography (CT) demonstrated a right sacral mass with posterior bony destruction. No local regional lymphadenopathy was appreciated. Magnetic resonance imaging (MRI) revealed a 7.8 x 8.6 cm heterogeneously enhancing mass with a large soft tissue component (Figure 1).
Figure 1: Tumor imaging. (A). Axial and (B). sagittal T2 MRI of the pelvis demonstrating an intermediate to brightly heterogeneously enhancing mass in the right sacrum and soft tissue with boney destruction. CT-guided core needle biopsy showed a pigmented malignant spindle cell neoplasm. The main histological differential diagnosis was metastatic melanoma, primary CNS melanoma and MMNST. Positron-emission computed tomography (PET/CT) demonstrated that the sacral mass was centered on the right S2 nerve root, with anterior and posterior soft tissue extensions, and a large lytic component in the right posterior ilium. There was no evidence of metastatic disease. A trans-abdominal, stage I procedure for high sacrectomy, with colectomy and colostomy was performed. This was followed one week later by en bloc resection and sacrectomy with L3-pelvis posterior spinal instrumentation and fusion, and flap reconstruction. Intraoperative pathology revealed a pigmented spindle cell, malignant tumor involving the surgical margin. Additional tissue was excised with grossly negative margins.
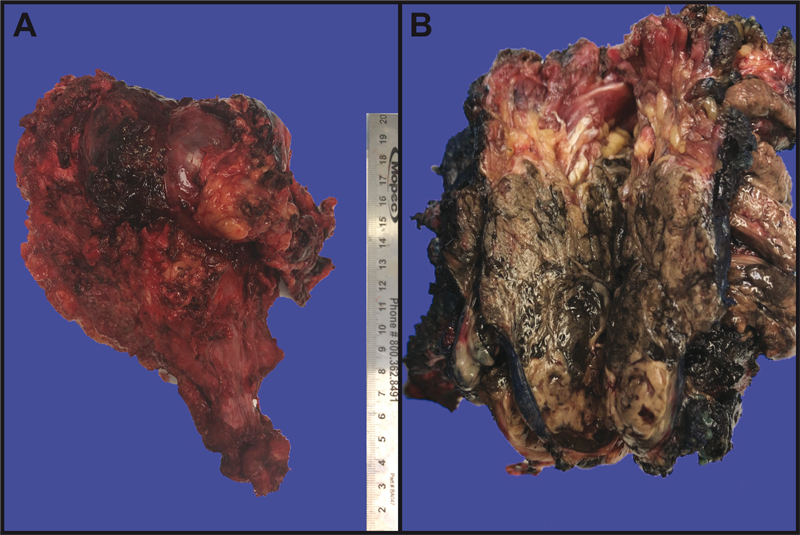
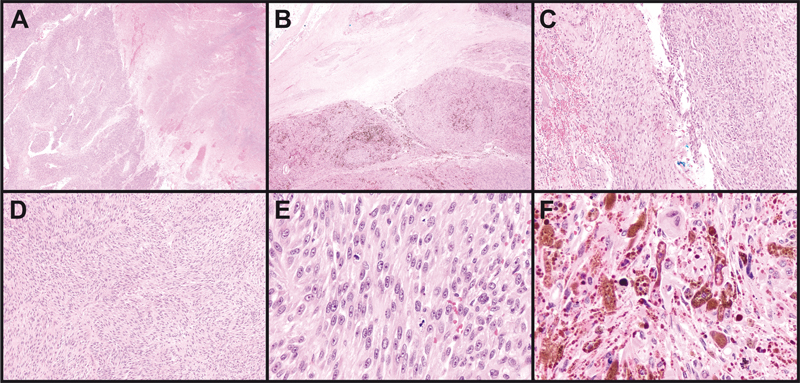
Figure 2: Gross sacrectomy specimen. (A). Lateral view, displaying a well-circumscribed tumor involving soft tissue and bone. (B). After inking the specimen blue, sectioning the tumor revealed the typical black “dried tar” appearance of cut surfaces. Pathology The resected tumor was 9.0 x 8.5 x 7.0 cm in dimension, centered in soft tissue, and eroded into adjacent bone. It was relatively circumscribed, heavily pigmented with a “black tar” appearance, and partly covered by a thin fibrous membrane. (Figure 2A-B). Microscopically, the cells were arranged in sheets, lobules, and fascicles (Figure 3A-D). Click here to view the full virtual slide. Large areas of necrosis were present (Figure 3A). Most tumor cells were spindle- to epithelioid-shaped with oval nuclei, vesicular chromatin, and prominent nucleoli (Figure 3E). Up to 18 mitotic figures were counted in ten 400X fields. Scattered melanin was identified within tumor cells and melanophages (Figure 3B and 3F). Focal areas showed Verocay-like nuclear palisading. Some areas contained highly pleomorphic cells demonstrating eosinophilic intranuclear pseudoinclusions (Figure 3F). The tumor was surrounded by a pseudocapsule containing neurofilament-immunoreactive normal ganglion cells and nerve tissue (Figures 3B-C and 4A). Multiple areas of pseudocapsular and lymphovascular invasion were noted. Occasional psammoma bodies were seen within the pseudocapsule adjacent to the tumor.
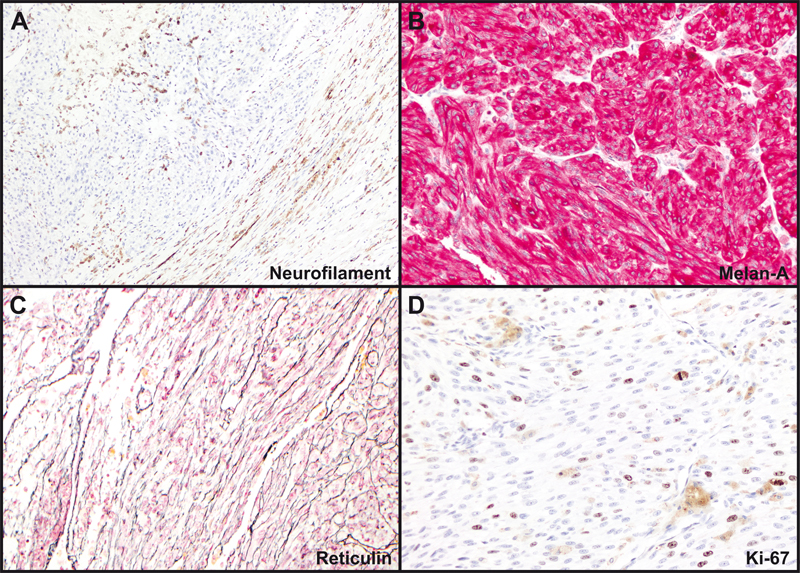
Figure 3: Tumor histology. (A). Geographic necrosis comprised approximately 30-40% of sampled tumor areas. (B). The tumor was lobular, pigmented, and surrounded by a pseudocapsule containing benign nerve elements. (C). The pseudocapsule incorporated benign ganglion cells (left). Some tumor areas were deceptively bland (right). (D). Most tumor areas contained spindled cells in a fascicular and vaguely nodular arrangement. (E). Nucleoli were prominent and mitoses were abundant. (F). Some areas were highly pleomorphic and/or demonstrated nuclear pseudoinclusions. Magnification: A-B, 20X; C-D, 100X; E-F 400X. Tumor cells were diffusely immunopositive for SOX10, HMB-45, and Melan-A (Figure 4B). S100 was positive in scattered cells. Cytokeratins AE1/AE3, BRAF-V600E, and PAX-8 immunostains were negative. INI1 (SMARCB1) was positive in tumor cell nuclei. Reticulin stain diffusely highlighted spindle tumor cell-associated basement membranes and showed focal lobular staining of small groups of epithelioid cells (Figure 4C). Ki-67 labeling was 5-10% in most areas and up to 20% focally (Figure 4D). PD-L1 was detected by immunohistochemistry (clone ZR3; PD-L1 expression score 2, detection cut-off score ≥ 1).
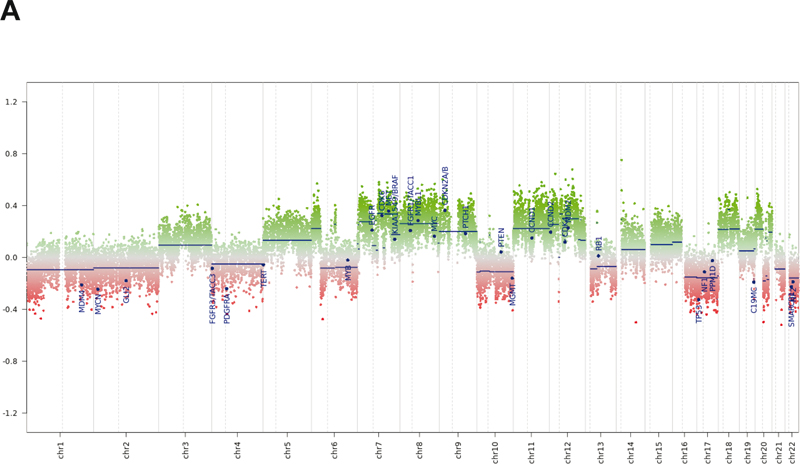
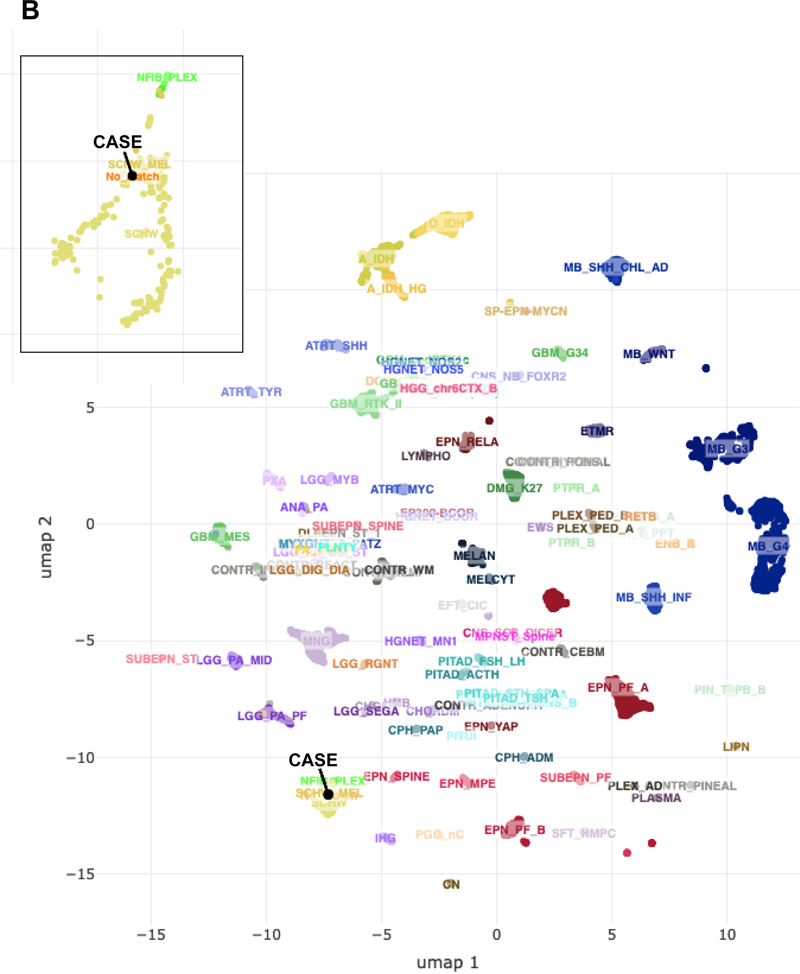
Figure 4: Tumor immunohistochemistry and reticulin stain. (A). Neurofilament- immunoreactive entrapped nerve elements within the tumor (upper left) and ganglion cells and axons within the pseudocapsule (lower right). (B). Diffuse positivity for Melan-A. (C). Pericellular and lobular reticulin staining. (D). Ki-67 immunohistochemical staining. Magnification: A, 40X; B-D, 200X. Next generation sequencing (NeoGenomics NeoTYPE Analysis Discovery Profile) and fluorescence in situ hybridization (NeoTYPE Discovery Solid Tumor FISH Panel) were performed on formalin-fixed paraffin-embedded (FFPE) tumor. Two predicted inactivating mutations were detected: a lysine methyltransferase 2c (KMT2C) splice site mutation (c.7443-1G>T) and a PRKAR1A frameshift mutation (c.1055del, p.R352Hfs*89). Additionally, a pathogenic missense mutation (c.548G>T, p.R183L) was identified in the G protein subunit alpha q (GNAQ) [7]. Missense variants of unknown significance were detected in AKT2 (c.1013T>G, p.V338G), KMT2C (c.7015A>G, p.N2339D) and SETD2 (c.65C>G, p.T22S). NF1 gene mutation was not detected. FISH demonstrated additional copies of BRAF (>2F, 52.0%; negative <25.1%) and MYC (3F, 30.0%; negative <16.2%), consistent with gains of BRAF (7q34) or chromosome 7 and MYC (8q24) or chromosome 8. Genomic DNA methylation analysis was performed on FFPE tissue using the Illumina Infinium Methylation EPIC BeadChip 850K microarray [8]. Methylation data was uploaded to the German Cancer Research Center (DKFZ) methylation-based classification platform for central nervous system tumors (www.molecularneuropathology.org). Results did not match any existing tumor methylation class (calibration scores < 0.9). Chromosome copy number analysis confirmed chromosome 7 and 8 gains, as well as gains of 6p, 6q, 11, 12, 18, and 20p, and loss of 1p, 16q, 17, 20q, and 22q, among other changes (Figure 5A). Uniform manifold approximation and projection (UMAP) dimensional reduction analysis comparing the tumor methylation data to the DKFZ brain tumor reference cohort [8] placed the tumor very near, or within, the schwannoma/melanotic schwannoma group (Figure 5B).
Figure 5: Genomic DNA analysis. (A). Chromosomal copy number data. (B). Unsupervised UMAP dimension reduction analysis of tumor genomic DNA methylation data. Although the tumor did not match a specific tumor methylation class using the DKFZ classifier, it grouped near, or with, schwannoma/melanotic schwannoma by UMAP (inset). Adjuvant therapy and Follow-up: Postoperatively, the patient received 5940 cGy radiation in 33 fractions to her pelvis. She then received three cycles of pembrolizumab. The fourth cycle was withheld due to autoimmune hepatitis. The patient had a dramatic improvement in pain from a visual analogue scale (VAS) pain score of 8-10/10 to 2-3/10. Her Karnofsky performance score was 90. CT scans five months after surgery demonstrated a new enhancing lesion of the pericardium, new gluteal mass, and new lytic lesions in the T2 and L2 vertebra and the left acetabulum. Positron-emission tomography (PET) showed these and multiple rib lesions. Core needle biopsy of the right gluteal muscle confirmed tumor recurrence. Six months after surgery, immunotherapy with ipilimumab and nivolumab was initiated. She completed her fourth cycle three months later. CT showed new and worsening osseous metastases, as well as innumerable sub-centimeter pulmonary metastases, and enlargement of the pericardial mass. Spinal MRI revealed a pathologic fracture of T2. The patient was hospitalized with right-sided radiculopathy and underwent surgical stabilization of T2 followed by radiation to residual T2 tumor. She also developed a 3.6 cm right frontal calvarial lesion, which was irradiated. The patient was then treated with doxorubicin. She had further progression at C2 three months after spinal surgery. The patient elected for hospice care and died one month later (18 mo after primary resection). Discussion MMNST are slightly more common in females (1.4:1 female to male) [3]. They are most often located paraspinally or in the gastrointestinal tract, but also occur at other sites [9, 10]. Their average age of presentation is 33.2 years for sporadic tumors and 22.5 years within the Carney complex. Grossly, MMNST are typically solitary, partially circumscribed or encapsulated, and heavily pigmented [2]. They range from 0.5 cm to 25 cm in diameter, but most exceed 5 cm [10]. MMNST are associated with nerves or soft tissue and may erode bone. True intraosseous examples are rare [11]. Microscopically, they are comprised of short fascicles or sheets of polygonal or spindled cells with a syncytial appearance. Vague tumor cell palisading or whorling may be evident [2]. Pigment is variable. Nuclei are typically round to ovoid with nuclear grooves, pseudoinclusions, and/or prominent nucleoli. Marked pleomorphism and nuclear hyperchromasia may be present [12]. Associated vessels are usually capillary-like. Psammoma bodies are present in ~50% of cases. Psammomatous variants may contain lipoma-like fat accumulation and occur more often in Carney complex [6, 11]. Residual ganglion cells may be identified in paraspinal examples. MMNST usually strongly express S100 and melanocyte markers (SOX10, HMB45, Melan-A, and tyrosinase) [13]. Although, examples of S100 patchy-positivity or S100-negative tumors have been reported [2, 13, 14]. Ultrastructurally, tumor cells exhibit both Schwann cell and melanocyte features, i.e., elaborate cytoplasmic processes, premelanosomes, and melanosomes [2, 10, 12]. Mitotic rate is the only histologic feature found to be predictive of clinical outcome. In one study, a mitotic rate of >2/10 HPF correlated with metastases (P=0.008) [34]. However, >50% of tumors that eventually metastasized did not show increased mitoses. MMNST are biologically distinct from conventional schwannoma. MMNST tend to occur in posterior spinal nerves and ganglia, while schwannomas additionally may involve other nerves, including cranial nerves. The peak age of MMNST onset is a decade younger than for schwannoma [12]. Unlike the latter, MMNST generally lack Antoni A and B regions and hyalinized blood vessels histologically. Gene expression profiling has shown significant differences between MMNST, melanoma, and schwannoma [13]. MMNST exhibited downregulation of genes involved in Schwann cell function, e.g., PMP22, PMP2, and MPZ, while genes related to melanin synthesis were upregulated in MMNST compared to schwannoma. In contrast to MMNST, conventional MPNST usually arise from peripheral nerves or extraneural soft tissue and are commonly seen in neurofibromatosis type 1 (NF1) [2], while MMNST only rarely occurs in NF1 [10, 15]. MPNST histologic features include monomorphic spindle cells with broad, intersecting, herringbone fascicles, alternating hyper- and hypocellular areas, a high mitotic count, and geographic necrosis [2]. MPNST also generally lack pigment and are negative for melanotic markers. Reported gene alterations in MPNST include NF1, CDKN2A, CDKN2B, TP53, TYK2, EED, and SUZ12 mutations and deletions, and EGFR, PDGFRA, and MET amplification [2, 16]. The uncommon epithelioid variant of MPNST arises from a pre-existing schwannoma and harbors SMARCB1 mutations [9, 11], whereas SMARCB1 has been reported to be intact in MMNST [13]. Distinguishing MMNST from melanoma can be difficult. Both may demonstrate spindled or epithelioid cells with pigment, violaceous macronucleoli, and positivity for S100 and melanocytic markers [9]. A lower Ki-67 labeling index may be suggestive of MMNST [2]. Extensive collagen IV or reticulin basement membrane staining supports Schwannian differentiation. A lobular/clustered tumor cell arrangement with syncytial-like cytology, psammoma bodies, and fat accumulation may also help distinguish MMNST from melanoma [9]. The genetics of MMNST are not completely defined. The majority of tumors are sporadic. PRKAR1A mutations and loss of PRKAR1A protein expression are seen in most cases and should prompt a search for other findings of the Carney complex [3, 4]. A complex karyotype including 22q monosomy, recurrent losses involving chromosomes 1, 2, 21, and 17p, trisomy 6p, and ring chromosome 11 have been described in MMNST [3, 4, 10, 17]. Melanoma typically shows 6p and 8q gains and 11q loss [17, 18]. MPNST exhibits gains of chromosomes 2, 7p, 8q, 14 and 17q, and loss of 9p, 11q, 13q, 17p, 18 and 22q [11, 19]. Schwannoma also shows 17p and 22q loss. This case thus exhibits reported karyotypic changes overlapping MMNST, MPNST, and schwannoma (8q gain, and 1p, 17p, and 22q loss), and MMNST and melanoma (6p and 8q gains) (Figure 5A). The case also demonstrates gene aberrations not previously ascribed to MMNST, namely, BRAF copy number gain and KMT2C splice site, and GNAQ R183L missense mutations. BRAF V600E mutations, not gains, are found in cutaneous melanoma and pigmented epithelioid melanocytoma [20]. Notably, the latter also may harbor PRKAR1A aberrations [21] or loss of heterozygosity [22]. GNAQ codon Q209 mutations are reported in leptomeningeal melanocytic neoplasms, uveal melanoma, and pigmented epithelioid melanocytoma [20, 21, 23, 24]. GNAQ variants have also been described in a malignantly transformed schwannoma (GNAQ T96S) [25] and an NF1-associated MPNST (GNAQ Y101X stop gain) [16] (Angela Hirbe, personal communication, July 2022). Therefore, unlike previously suggested for MMNST [2, 23], neither GNAQ nor PRKAR1A mutations can be relied on to differentiate MMNST from melanocytic lesions. Although, PRKAR1A and GNAQ mutations coexist in the MMNST described herein, we found no other documentation of their coexistence in the entities discussed above [16, 20, 21, 23, 24, 25]. However, in an important study comparing MMNSTs to melanocytic lesions, only exon 5 of GNAQ containing the Q209 codon was sequenced [23]. Thus, in that study GNAQ codon T96, Y101, and R183 hotspot mutations within exons 2 and 4, respectively, could not have been detected. GNAQ mRNA tissue distribution is greatest in the brain [26]. A propensity for GNAQ mutations in neoplasms of other neuroectoderm/neural crest-derived tissues, i.e., melanocytes and nerves, is thus not surprising. However, GNAQ is also important in vascular development. GNAQ R183L occurs in brain endothelial cells of Sturge-Weber syndrome [27]. GNAQ R183L and R183Q mutations have been identified in capillary malformations [7, 28, 29] and GNAQ Q209 mutations in other benign vascular lesions [30]. Additional tissues also highly express GNAQ and GNAQ mutations may occur in endocrine tumors and other neoplasms [31]. Achieving long-term survival in MMNST is hampered by its tendency to metastasize and recur, and its propensity to involve the spine or other structures, which preclude complete surgical resection [32]. One study reports a 20-year remission rate of 67% with total resection [33]. However, Torres-Mora et al [13] reported MMNST local recurrence and metastatic rates of 35% and 44%, respectively (mean follow-up 55 mo), with 73% of metastases occurring in <4 years. Metastases are typically to the lung and pleura. Radical resection with or without radiation and/or chemotherapy is the mainstay of treatment. Although, radiation has not shown a clear benefit [12]. A few reports using traditional chemotherapeutics, e.g., carboplatin and etoposide, showed low response rates and no survival benefit [14]. While immune checkpoint inhibitors are commonly used in cutaneous melanoma, their use in MMNST is not well-established. This patient with documented PD-L1 expression was treated with the checkpoint inhibitors pembrolizumab, ipilimumab, and nivolumab with pain improvement, but no objective response in disease progression. Bajpai et al., however, described a patient with recurrent and metastatic MMNST who underwent multiple surgeries, external beam radiation, and nivolumab treatment [34]. Ipilimumab was added after disease recurrence. The authors reported a survival of 51 months from initial diagnosis and 35 months after the start of checkpoint inhibitor therapy. Vining et al. described a patient with retrocaval MMNST who experienced decreased pain and disease stabilization with pembrolizumab prior to resection [35]. Thus, the use of immune checkpoint inhibitors in MMNST is limited to three reported cases, nevertheless, they suggest some evidence that they may provide symptomatic improvement and clinical response in some PD-L1-positive MMNST patients. References
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |