|
|
|
Free Neuropathology 3:8 (2022) |
|
Review |
|
Neurodevelopmental disorders: 2022 update |
|
Miguel Sabariego-Navarro1, Álvaro Fernández-Blanco1, Cesar Sierra1, Mara Dierssen1,2,3,4 |
|
1 Center for Genomic Regulation, The Barcelona Institute for Science and Technology, Barcelona, Spain |
|
Corresponding author: |
|
Submitted: 09 February 2022 Accepted: 08 March 2022 Copyedited by: Christian Thomas Published: 21 March 2022 |
|
Keywords: Mitovesicles, Repeatome, tRNAs methylation, Chromatin remodeler, Autism spectrum disorder, Aicardi-Goutières syndrome |
|
Abstract With a prevalence of 2-4% of the worldwide population, neurodevelopmental disorders (NDDs) comprise a heterogeneous group of disorders associated with neurodevelopmental dysfunction, including intellectual disability (ID), autism spectrum disorder (ASD), Down syndrome (DS) and attention-deficit/hyperactivity disorder (ADHD) among others. However, due to their heterogeneity and overlapping clinical features, NDDs such as ASD are often misdiagnosed, while for others with more distinct symptoms, such as Rett syndrome or DS, the mechanisms underlying their pathogenesis remain elusive. Last year, important steps in the mechanistic understanding of several NDDs have been achieved. New preclinical models demonstrated causality between PAK3 mutations and disorders associated with social deficiencies. ARID1B mutations have been linked to neuroectoderm specification in Coffin-Siris syndrome and DNA damage was established as an important pathologic mechanism in Aicardi-Goutières syndrome. Moreover, alterations in basic molecular processes including translation and histone acetylation have been established as major traits in the pathology of X-linked ID and Rett syndrome, revealing new pathogenetic mechanisms. Last year, advances in bioinformatics have begun to shed light on the human repeatome, a largely unexplored part of our genome, and how alterations in these sequences have a central role in ASD. The role of mitochondria in neuropathology was clarified last year with the discovery of previously unknown vesicles derived from mitochondria with a putative role in DS. An interesting discovery in the field of basic neurodevelopment showed that during postnatal brain development, changes in genome architecture and transcriptional dynamics progress independently of sensory experience. Finally, our neurocentric views of NDDs are changing as new players such as astrocytes are revealed to be crucial in neuropathology. The role of astrocytes has been clarified for some pathologies such as ASD and DS, linking well-known genetic mutations to impaired astrocyte function. Abbreviations ADAR1 - double-stranded RNA-specific adenosine deaminase, AGS - Aicardi-Goutières syndrome, ASD - autism spectrum disorder, ATP - adenosine triphosphate, cGAS - GMP-AMP synthase, cKO - conditional knockout, Dip-C - diploid chromatin conformation capture, DNA - deoxyribonucleic acid, DS - Down syndrome, EV - extracellular vesicles, GABA - gamma-aminobutyric acid, γH2AX: H2A - histone family member X, hiPSC - human induced pluripotent stem cells, Iba1 - ionized calcium-binding adaptor molecule 1, ID - intellectual disability, IFN - interferon, IP3R2 - 2 inositol 1,4,5-trisphosphate receptors, LC-MS - liquid chromatography-mass spectrometry, MeCP2 - methyl CpG binding protein 2, NDD - neurodevelopmental disorder, RNA - ribonucleic acid, RNASEH2 - ribonuclease H2, NMDA - N-methyl-d-aspartic acid or N-methyl-d-aspartate, NOVA1 - neuro-oncological ventral antigen 1, PSD95 - postsynaptic density 95, RTT - Rett syndrome, RPS4Y1 - 40S ribosomal protein S4, TADs - topologically associated domains, TRs - tandem-repeat sequence, TREX1 - three prime repair exonuclease 1, tRNA - transfer RNAs, XLID - X-linked intellectual disability. Introduction Single-term search in the PubMed database with the search term “neurodevelopmental disorders” from January 1st to December 31st 2021 retrieved more than 10,000 papers. In this review, we selected the 10 most interesting discoveries in the field during the last year. Instead of centering our efforts around papers published in the highest impact factor journals, we decided to select the discoveries that open new research avenues and that can be expected to have substantial implications in the study of neurodevelopmental disorders paving the way to a change of paradigms. The major steps made during 2021 comprise very different topics, from the discovery of new shared molecular mechanisms involved in several neurodevelopmental syndromes such as Aicardi-Goutières syndrome or Coffin-Siris to disentangling the complexity of autism spectrum disorders by the identification of new players at the network, on the cellular and the molecular levels. Advancement in modeling tools holds the promise to not only increase our understanding of the origin of neurodevelopmental disorders, including their evolutionary aspects, but also to start proving causality. In the last year, the role of several underexplored biological underpinnings such as tRNAs methylation or mitochondrial-derived extracellular vesicles were clarified in neurodevelopmental disorders, opening new perspectives for diagnosis and treatment. Finally, new tools appeared to study human neurodevelopmental disorders, partially solving some of the associated problems with the use of animal models. The final list of papers and topics of 2021, selected as very interesting and/or important for the field of neurodevelopmental disorders, comprises:
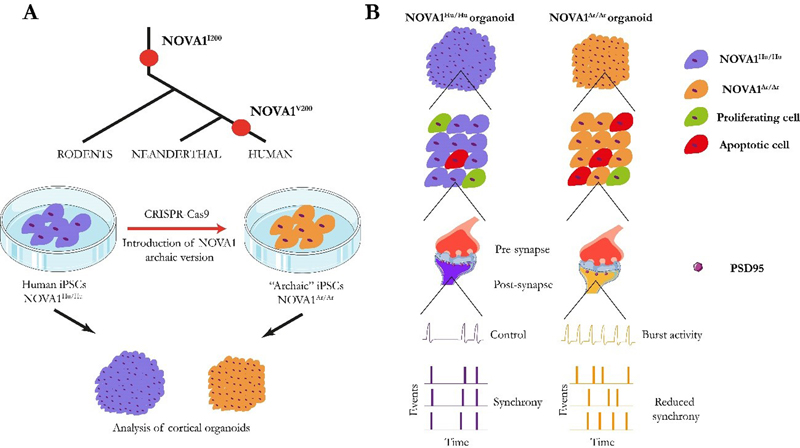
Reconsidering the molecular neuropathological basis of Aicardi-Goutières syndrome (AGS) Aicardi-Goutières syndrome (AGS) is a rare neurodevelopmental disorder. The main neuropathological signs are progressive microcephaly associated with basal ganglia and white matter calcifications, leukodystrophy, cerebral atrophy, and variable increase of lymphocyte count in the cerebrospinal fluid. It is caused by recessive mutations in nine known genes identified until now, namely TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, MDA5, LSM11 and RNU7-1 that result in upregulation of type I interferon (IFN) signaling and is accompanied by neuroinflammation [1]. Increased type I IFN activity is detected in cerebrospinal fluid and serum, and increased expression of IFN-stimulated gene transcripts in peripheral blood. Moreover, a positive correlation between the level of cerebrospinal fluid IFN activity and the degree of motor and intellectual disability has been reported [2]. Astrocytes are the main cell type that produce IFN-α in the brain and magnetic resonance image studies in patients suffering AGS show calcifications in the white matter and white matter signal changes in the frontotemporal regions with associated astrocytosis [3]. Interestingly, AGS-related genes are critical in nucleic acid metabolism function [4, 5], and involved in the removal of redundant endogenous or exogenous DNA, RNA, or DNA/RNA hybrids. It is assumed that failure to remove intracellular remnants of nucleic acids leads to stimulation of the innate and adaptive immune responses, so that type I interferonopathies would represent a failure of self- versus non-self-discrimination, as these essential antiviral systems can also be triggered by host DNA and RNA. The ribonuclease H2 (RNASEH2), linked to AGS, is a genome surveillance factor, which has the role of constantly inspecting DNA integrity and removing ribonucleotides that incorporate by error into replicating DNA [2]. However, the role of IFN hyperactivity and RNASEH2 mutations and their possible functional interaction on AGS neuropathology is poorly understood. Although the neurotoxic potential of type I IFN was highlighted by studies that recapitulated the neuropathological features of AGS in transgenic mice chronically producing IFN-α from astrocytes [6], available genetic AGS mouse models fail to show type I IFN response and inflammation in the brain [7, 8]. Now, Aditi et al. have provided evidence that DNA damage-dependent signaling rather than type I IFN signaling underlies neurodegeneration, in this class of interferonopathy. They generated a murine model of neural Rnaseh2b inactivation (Rnaseh2bNes-cre) [9]. These mice showed smaller cerebellar volume and reduced granule cell and interneuron number. The authors found that reduced cerebellar volume was explained by the loss of proliferative neurons as a consequence of DNA damage-induced cell death. They found increased DNA double-strand breaks in cells using the H2A histone family member X (γH2AX) marker. The authors used RNA-sequencing to compare cerebellar tissue of Rnaseh2bNes-cre and controls. They found that genes involved in type I IFN-α response pathway were particularly enriched in Rnaseh2bNes-cre cerebellar cells indicating that RNASEH2 deficiency leads to an increased type I IFN response. Consistent with the upregulation of neuroinflammatory genes in Rnaseh2bNes-cre cerebellum, they also found astro- and microglial genes such as glial fibrillary acidic protein (Gfap) and ionized calcium-binding adaptor molecule 1 (Iba1) were particularly upregulated in Rnaseh2bNes-cre cerebellum. These data suggest an ongoing astrocytosis and microglial activation that are hallmarks of neuroinflammation. Remarkably, inactivation of tumor protein p53, which induces apoptosis in damaged cells, rescued DNA damage-induced cell death and cerebellar atrophy but led to accumulation of cytoplasmic DNA damage that resulted in the development of medulloblastoma. The authors also demonstrated that neurodegeneration of Rnaseh2bNes-cre mice was not driven by type I IFN signaling since the deletion of cyclic GMP–AMP synthase (cGAS), a factor that is required for type I IFN activation, led to similar phenotypes in Rnaseh2bNes-cre mice. All these data together provide new levels of understanding about AGS neuropathology and suggest that neuroinflammation is not the main cause of AGS phenotypes, but is a consequence derived from increased DNA damage due to Rnaseh2b mutations. In this regard, those individuals with additional mutations in DNA repair genes might be more prone to present exaggerated immune responses as a consequence of increased DNA damage. This discovery constitutes a paradigm-shift in interferonopathies and changes the assumptions about the genetic and molecular causes of AGS. Reconsidering the leading cause of AGS neuropathology can help not only to disentangle the complex neuropathology of this neurodevelopmental disorder, but also to specifically design strategies that would help to ameliorate the symptoms associated with DNA damage-driven neuroinflammatory response in AGS. Understanding cortical neurodevelopment from evolution Genetic disease is a necessary product of evolution. In fact, evolutionary pressures have established the risk for many neuropsychiatric or neurodevelopmental diseases. Nearly all genetic variants that influence disease risk have human-specific origins. Fundamental biological systems, such as DNA replication, transcription and translation, evolved very early in the history of life, to give rise to cellular life, but also created the potential for disease. Among those, alternative splicing is a form of genetic regulation that enables the production of multiple proteins from a single gene. Differences in splicing arose rapidly during a recent evolutionary transition and appear to contribute to adaptation but also to brain disorder. It is known that the transcriptional machinery is often mutated in neurodevelopmental disorders [10]. The brain displays the most complex pattern of alternative splicing of the body [10], with neuron-specific alternative exons such as DNA-binding proteins and histone modifying enzymes that regulate evolutionarily conserved transcription. Given that alternative splicing is necessary for the proper cortical development [11, 12], a better understanding of how mutations in genes associated with alternative splicing would help to unravel the enormous complexity of several NDDs. One interesting example is the neuro-oncological ventral antigen 1 (NOVA1), which encodes a RNA-binding protein that acts as a brain-specific splicing factor. It regulates neuronal alternative splicing of genes responsible for synapse formation in the developing nervous system [13]. Increasing evidence has implicated NOVA1 in numerous pathological processes and neurodevelopmental disorders [14, 15]. Mutations in NOVA1 have been associated with impairment in the development of the motor system [16] and severe neurodevelopmental delays [17]. Last year, a comprehensive analysis of genetic variation between modern humans and Neanderthals performed by Trujillo et al. detected a specific NOVA1 genetic variation [18]. Even though the genomes of humans and Neanderthals are very similar, this archaic NOVA1 version could help to explain how NOVA1 variants can differentially regulate alternative splicing during human brain development. To study the functional relevance of NOVA1 mutations, authors used CRISPR-Cas9 genome-editing technology to introduce the archaic variant into the genome of human induced pluripotent stem cells (hiPSCs). hiPSCs carrying the archaic mutation (NOVA1Ar/Ar) and control hiPSCs (NOVA1Hu/Hu) were used to generate cortical organoids to investigate the impact of the NOVA1Ar/Ar variant on the human brain. Trujillo and colleagues found that NOVA1Ar/Ar cortical organoids were smaller than NOVA1Hu/Hu, a phenotype that was explained by a reduced number of rosettes and a higher number of apoptotic cells in NOVA1Ar/Ar along with a reduced number of proliferating cells compared to the NOVA1Hu/Hu organoids. The authors also found several genes differentially expressed in NOVA1Ar/Ar such as RPS4Y1 and NNAT, which are involved in neuronal differentiation, TDGF1, involved in cell migration and proliferation, or PAX6 and LHX5, transcription factors associated with cell differentiation during brain development. Trujillo and colleagues investigated the gene-splicing variants of NOVA1Ar/Ar and NOVA1Hu/Hu using single-nuclei RNA-seq and identified differences in alternative splicing events affecting 122 genes. Remarkably, most of the differentially spliced genes in NOVA1Ar/Ar organoids are involved in synaptogenesis and neuronal connectivity. More in detail, NOVA1Ar/Ar cells had reduced levels of pre- and postsynaptic markers such as PSD95 and NMDA, closely related with activity-dependent plasticity. Finally, they evaluated how these synaptic changes could impact neuronal network activity using a multielectrode array that measures several electrophysiological properties. NOVA1Ar/Ar cells showed increased number of bursts, lower synchrony and increased variability compared with NOVA1Hu/Hu organoids (Fig. 1). Overall, their results suggest that the archaic NOVA1 variant causes changes in alternative splicing of genes involved in brain development, proliferation and synaptic plasticity that might explain how mutations of the NOVA1 gene can contribute to different alterations in NDDs. Specifically, NOVA1 would negatively impact neuronal proliferation and survival, but also synaptic connectivity as a result of the reduced expression of proteins important for activity-dependent plasticity. This new study helps disentangle the evolutionary origins of NDDs, also pointing to relevant biological processes.
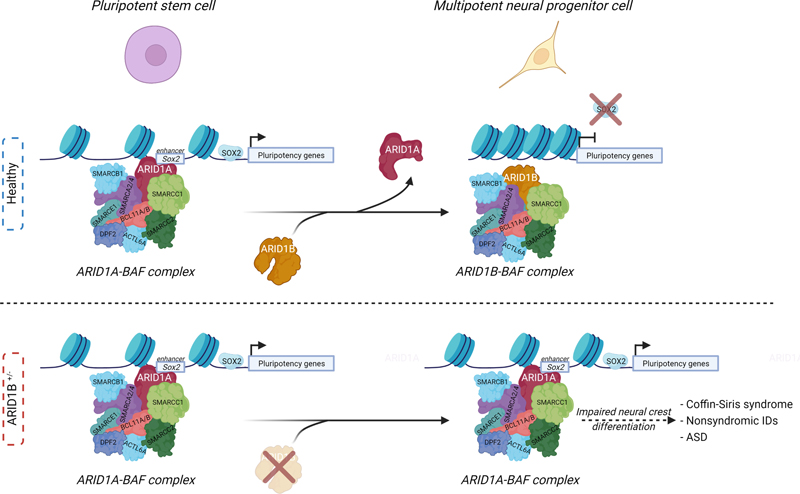
Fig. 1. Schematic representation of how the archaic NOVA1 variant affects cellular, molecular and neuronal network activity in cortical organoids. A) NOVA1 archaic version was introduced in iPSCs derived from humans. NOVA1Hu/Hu and NOVA1Ar/Ar hiPSCs were differentiated into cortical organoids and their cellular, molecular and physiology were studied. B) Size of NOVA1Ar/Ar organoids was smaller than NOVA1Ar/Ar organoids due to decreased neuronal proliferation and increased apoptosis. Single-nuclei RNA-seq revealed reduced levels of synaptic proteins such as PSD95. The study of electrophysiological properties by means of a multielectrode array revealed an increased burst activity in NOVA1Ar/Ar cells along with higher variability and reduced levels of synchrony when compared to NOVA1Hu/Hu. New 3D in vitro models to unveil neuropathological mechanisms of human neurodevelopmental disorders Until recently, the study of neurodevelopmental disorders (NDDs) has mainly relied on in vivo animal models to characterize the changes occurring during early brain development. However, the establishment of in vitro models of brain development derived from hiPSCs represents an unprecedented opportunity to study key brain developmental processes, such as neuronal cell morphogenesis, migration and connectivity in human tissue. Although this field is still in its infancy, an increasing number of studies demonstrate its potential to shed light on the mechanisms underlying NDDs. This potential is illustrated by recent advances in the study of Rett syndrome (RTT), a NDD characterized by cognitive, speech, behavioral and motor problems, that starts in the first year of life and that predominantly affects females. Rett syndrome is a rare and complex X-linked neurodevelopmental disorder associated with mutations in methyl CpG binding protein 2 (MeCP2) that cause its loss of function. MeCP2 is an epigenetic reader that binds to methylated DNA to promote transcriptional repression. Male homozygous MeCP2-/Y mice are the most frequently used RTT model due to the early development of severe phenotypes, but even though the use of these models has significantly contributed to the understanding of the mechanisms underlying RTT, their clinical relevance might be limited. In fact, their face validity is constrained as male mice are used to model a disease mainly affecting females, and female patients have a milder phenotype than the one observed in mice. To overcome these limitations, several methodologies have been established to model RTT with hiPSCs from patients by developing 2D-based neuronal differentiation protocols. More recently, the generation of 3D human brain organoids, a more complex approach that better recapitulates human neurodevelopment in vitro, has been established. These culture platforms are increasingly contributing to a better understanding of the RTT etiology. The tremendous potential of such methodologies is illustrated by the recent work of Samarasinghe et al. [19] who showed that brain organoids derived from induced pluripotent stem cells from individuals with RTT reproduce patterns of electrical brain activity that resembled seizures, a hallmark of the condition. Using calcium sensor imaging and extracellular recording approaches, they demonstrated highly abnormal and epileptiform-like activity, accompanied by transcriptomic differences revealed by single-cell analyses. Interestingly, Trujillo et al. [20] used an innovative pipeline to model RTT neurodevelopment demonstrating that neurons differentiated from human MeCP2-KO hiPSCs show altered expression of synapse-relevant genes, synaptic morphology, and decreased calcium and network activities compared to control neurons. An in silico neural network simulation affirmed that synaptic structural parameters link with neural network activity, supporting the approach of rescuing synaptic structure and increasing activity. They differentiated MeCP2-KO hiPSC into in vitro models of increasing complexity. First, 2D monolayer neuronal cultures revealed an impaired neurotransmission. The same hiPSC were then used to produce a novel system of 3D brain neurospheres derived from a 50/50 mixture of healthy and MeCP2-KO hiPSC to better mimic the mosaicism observed in female patients, a phenomenon produced by random X-inactivation. This new model will be of great utility to investigate RTT and other X-linked genetic conditions. As a last step, complex 3D MeCP2-KO cortical organoids were also produced. Interestingly, this pipeline revealed important features of RTT pathology. The researchers found that RTT organoids displayed smaller diameters and both MeCP-KO neuronal cultures and organoids showed that synaptic and neurotransmitter pathophysiology principally concentrated in glutamatergic and cholinergic dysregulation. Furthermore, Nefiracetam, a cholinergic, GABAergic, and glutamatergic agonist, and PHA 543613, a α7-nAChR agonist, had a rescuing effect supporting glutamatergic and cholinergic dysregulation. New insights and tools to explore the genome architecture and transcriptional dynamics in the mammalian brain development Neurodevelopmental processes rely on the orchestration of finely tuned gene expression programs, which depend on the regulation of chromatin structure. Over the last few years, mutations in genes encoding factors that can modify chromatin regulation and nuclear architecture have emerged as a frequent cause of NDDs. Post mortem studies on brains from patients with different NDDs found chromatin and other epigenetic alterations linked with abnormalities in neural development. This is the case of Opitz-Kaveggia and Fryns-Lujan syndromes, caused by mutations in MED12 [21], and Cornelia de Lange syndrome, which results from mutations in NIPBL or in the cohesin subunits coding genes SMC1 and SMC3 [22]. Mutations in the CTCF gene are also associated with intellectual disability [23, 24]. On the other hand, our knowledge of the critical role of the genome’s 3D organization in gene regulation is steadily increasing. This is important since the expression of a given gene in a specific time and cellular type is known to depend on its location within the nucleus and on its contacts with other loci, forming topologically associated domains (TADs). However, although previous studies have contributed to characterize the transcriptome and 3D genome in the brain, they failed to answer this question mainly due to the use of bulk chromatin conformation capture techniques, which can only assess averaged maps of the 3D genome. This constitutes an important limitation in a highly heterogeneous tissue such as the brain. Now, Tan et al. [25] have overcome this limitation thanks to the use of a new diploid chromatin conformation capture (Dip-C) method [26], a chromatin conformation capture method that allows to distinguish between the two haplotypes of each chromosome, which allowed them to create a 3D genome atlas of the developing mouse forebrain, including ~2,000 single-cell contact maps and ~800 3D structures from two brain regions and 6 time points. In parallel, they also produced a single-cell transcriptomic atlas to shed light on the “structure-function” relationship question during neurodevelopment. Importantly, this approach revealed a major post-natal transcriptomic and 3D structure reorganization. In other words, the gene expression and chromatin architecture profiles of postmitotic neurons continue to mature during postnatal development. At the 3D genome architecture level, the radial positioning (preference for the nuclear periphery or nuclear core) of genes was the most prominent difference between neonatal and adult neurons. A similar phenomenon was previously thought to be unique for certain gene clusters, such as olfactory receptors (OR), in olfactory receptor neurons [27] development. Importantly, during developmental stages, ORs genes move to the nuclear interior, detaching from the nuclear laminin, thereby allowing their activation, to ensure that each olfactory receptor neuron expresses one, and only one, OR, a phenomenon called the “one neuron-one receptor expression” [27]. These results, however, show that this behavior might represent a generalizable principle of neuron development not restricted to olfactory receptor neurons. Strikingly, Tan et al. also showed that these transformations of the 3D genome are independent of sensory experience, suggesting that the 3D transformation is genetically predetermined. Altogether, these results provide new tools and a conceptual framework to shed light on the molecular mechanisms governing neurodevelopmental processes at an unprecedented resolution. New cellular mechanisms and new tools to establish causality in ASD Throughout the last years, advances in the field of genetics have led to the discovery of hundreds of genes that contribute to serious deficits in communication, social cognition, and behavior of autism spectrum disorder (ASD). However, these genes only account for 10–20% of cases, and similar pathogenic variants may lead to very different levels of affectation. In fact, understanding causal relationships between genes and phenotypes remains a challenge. Recent evidence suggests that dysfunction of Rho family guanosine triphosphatases (Rho-GTPases) contributes substantially to the pathogenesis of NDDs [28, 29], and as many as 20 genes encoding Rho GTPase regulators and effectors are listed as ASD risk genes [30, 31]. Those genes are promising candidates to explain ASD neuropathology. Rho GTPase signaling is essential for cellular signaling and cytoskeleton dynamics [32, 33]. One interesting example is the p21-activated kinase 3 (PAK3), which is a downstream component of the Rho-GTPase cascade. Genetic studies in humans revealed that mutations of PAK3 are associated with moderate to severe intellectual disability (ID) and with synaptic alterations in post mortem human tissues [34-36]. Animal models with Pak3 mutations (mPAK3) also recapitulate these phenotypes and have shown that PAK3 is important for recognition memory [37, 38]. In fact, PAK activity, in combination with RAC (a Rho-GTPase), has been suggested to be responsible for the autistic cellular and clinical phenotypes in mouse models of ASD, and may be related to synaptic plasticity alterations [39]. However, a causal link between these mutations and autistic phenotypes has not yet been directly demonstrated. One possible problem is the lack of spatial-temporal resolution of the investigations. For example, a gene mutation may have specific neuropathological impact on a subpopulation of cells, which cannot be selectively targeted. This is the case with memories, which are believed to be stored in a subpopulation of neurons that get activated and modify their functional properties during learning. Those are called engram cells [40, 41], and might be specifically affected by PAK3 mutations. For this reason, Zhou et al. designed a tetracycline-inducible system (tTA/tetO) under the control of cFos, to express a mutant PAK3 form tagged with green fluorescent protein (mPAK3-GFP) specifically in neurons that were activated during social interaction [42]. cFos is an immediate early gene associated with neuronal activity and synaptic plasticity and is often used to identify engram cells [40, 41, 43]. The authors showed that disrupting PAK3 signaling by the expression of mPak3 in cFos-expressing neurons during social interactions in fact was sufficient to impair social memory. Contrarily, rescuing mPak3 levels in the same animals restored social memory deficits. The fact that Pak3 disruption in just a subset of cFos positive cells is sufficient to produce social memory impairment underscores the critical importance of increasing the resolution of the techniques we use. In this regard, an increasing number of studies are using single-cell omics for resolving the cell-specific impact of specific mutations, and single-cell RNA seq data are available from post mortem neurotypical human brain regions. Even though the mechanisms by which mPAK3 was altering social memory engrams were not covered in this study, to date this is the first experimental demonstration that establishes the necessity of PAK-dependent molecular changes in engram cells underlying social memory pathology. Certainly, this cell-specific system can be used to establish molecular causality of memory deficits in different mouse models of NDDs given the ability to fine-tune the expression of particular genes that could be potentially implicated in the neuropathology of these disorders in an inducible manner. Increasing the resolution and selectivity of our experimental approaches will certainly uncover new neuropathological mechanisms and possibly help to understand the actual contribution of risk genes to the pathogenesis of autism and other NDDs. New mechanisms in ASD pathology: astrocytes, ATP and GABAergic signaling The research in ASD has traditionally focused on neuronal dysfunction, as many ASD genes encode for proteins that have a role in synaptic function, thus making the views of the pathological features of ASD mainly “neurocentric”. However, selectively focusing on neuronal mechanisms has proven to be limited, and thus, alternative biological analyses may help to reveal previously unknown cellular and molecular mechanisms. Astrocytes are newly identified players in several brain pathologies, and could play an important role in ASD. In fact, RNA sequencing revealed a close association between ASD and the genes related to glial cell activation, immune and inflammatory categories [44]. Studies in post mortem brain tissue from donors affected by ASD have shown alterations in the expression of astrocyte markers, and iPSCs derived from ASD patients present alterations in astrocytic development [45, 46]. One recent preprint shows that astrocytes from ASD patients cause autistic phenotypes when engrafted in mouse pups, establishing an important milestone in the ASD field [47]. However, whether astrocytes alterations are the cause or the consequence of ASD and how this glial cell type contributes to ASD pathology remains unclear. Interestingly, de novo mutations in type 2 inositol 1,4,5-trisphosphate receptors (IP3R2) gene have been identified in ASD patients [48]. This receptor mediates the activation of astrocytes by increasing cytoplasmic calcium signaling [49], and previous studies in Ip3r2 knockout (Ip3r2 KO) mice showed selective dysfunction of astrocytes but not neurons leading to social behavior impairment, as revealed by increased repetitive behaviors and impaired social approach [50]. Wang et al. [51] now show that astrocyte calcium signaling dysfunction can result in ASD-like behavior in mice. The authors generated a conditional knockout (cKO) mouse for Ip3r2 using an astrocyte-specific promoter, Aldh1L1, that induces the Ip3r2 knockout during adulthood. This astrocyte specific Ip3r2 KO in adult mice was sufficient to produce autism-related behaviors, suggesting that these phenotypes are not just a consequence of altered neurodevelopment. To further investigate the mechanism underlying social behavior alterations and astrocyte dysfunction in Ip3r2 mutant mice, they analyzed the levels of different gliotransmitters. They found a strong reduction in ATP levels, a known gliotransmitter, in Ipr32 cKO mice. This reduction in ATP concentrations was only found in astrocytes but not in neurons isolated from Ipr32 cKO mice, indicating astrocytes-specific alterations. Using chemogenetic techniques and an ATP sensor under an astrocytic promoter, they demonstrated that ATP release was significantly reduced in Ip3r2 cKO and that ATP administration led to a total rescue of social interaction. This indicates the need of ATP release by astrocytes for proper social interaction. This finding establishes a new pathogenic mechanism: lower release of ATP after astrocyte activation leads to an increase of GABAergic inhibitory activity. However, the pathogenetic mechanisms are certainly more complicated, as shown by other recent studies reporting that astrocyte dysfunction could lead to an altered microglial state [52], and lead to increased neuroinflammatory processes. In conclusion, astrocyte activity, mediated by calcium signaling pathways, plays a pivotal role in ASD pathology. Disentangling the role of repeatome in ASD Approximately half of the human genome consists of repetitive DNA sequences, and is called the repeatome. Repetitive DNA consists of more than one million tandem repeats, sections of DNA in which a sequence is replicated many times in tandems, interspersed repeats and copy number variants, which biology remains under-studied and poorly annotated, being considered as “junk DNA” compared to the protein coding DNA [53]. However, more than 50 diseases such as Huntington disease, Fragile X, various ataxias and a major subset of amyotrophic lateral sclerosis and frontotemporal dementia cases are caused by an expansion of a tandem-repeat sequence (TRs), showing the importance of this non-coding DNA in some pathologies [54]. Repeat-associated non-ATG translation (RAN translation) of toxic peptides may contribute to the pathogenesis of those disorders and emerging evidence suggests that TRs can also regulate gene expression in healthy individuals [54]. Nonetheless, the role of TRs in polygenic pathologies such as ASD remains largely unexplored. Even though changes in the number of copies of large segments of DNA have been implicated in ASD pathology [55], the capacity to analyze alterations in TRs from a genome-wide point of view was, until now, unachievable. Mitra et al. have now developed a new bioinformatic tool capable of analyzing TRs in people with ASD and their family members [56]. This new bioinformatic tool that the authors named MonSTR, allowed the researchers to discover that TRs expansions are more common in people with ASD than in their unaffected siblings. The expansions occurred in genes involved in brain development such as FOXP1. Some genes previously associated with ASD, e.g. PDCD1 or KCNB1 present some tandem-repeat mutations, increasing the possibility that these repeats are involved in ASD pathology. In a recent paper, Trost et al. [57] also analyzed the role of TRs in ASD but with a different approach, not taking into account the appearance of de novo mutations. This study used three times more genomes as compared to the analysis of Mitra et al., thus substantially increasing the statistical power. Although the studies identified different mutations, probably due to the differences in their methodological approach, they were more complementary than controversial. Interestingly, the study from Trost et al. relates several TRs expansions with particular clinical features like cognitive disturbances. The findings open a new research line that could contribute to disentangle the contribution of TRs to other neurodevelopmental disorders and pathologies. However, the most critical question now is how these alterations in TRs generate disturbances in neurodevelopmental processes, accounting for the multifactorial nature of ASD with the environment playing a pivotal role, thus opening the question of how the environment interacts with these alterations in TRs. Key switch between chromatin remodeler BAF subunits explains Coffin-Siris syndrome neuropathology The brain exhibits an extraordinarily precise morphogenesis that leads to the correct cell types and proportions at the appropriate sites. During the neurodevelopmental processes that lead to the formation of the central nervous system, cell fate is acquired by the decline in neural stem cell self-renewal and amplification potential to give rise to fate-committed progenitors that will ultimately undergo differentiation into neurons (neurogenic) or glial cells (gliogenic) [58]. The establishment of these cell fates requires the orchestration of timely regulated gene expression programs which, in turn, depend upon epigenetic and chromatin regulators. ATP-dependent chromatin remodeling factors have been described to be vigorous regulators of chromatin state and DNA accessibility, both of which have an important impact on the coordination of gene expression. Interestingly, a substantial proportion of NDD associated genes are involved in chromatin and/or transcriptional regulation including the broad family of ATP-dependent chromatin remodelers including the multi-subunit BRG1/BRM associated factor (BAF) chromatin modifier family, also known as SWItch/Sucrose Non-Fermentable (mSWI/SNF) chromatin remodeling complex. A DNA-binding domain targets the SWI/SNF complex to specific genes and facilitates DNA access for transcription factors. These complexes play a central role in different developmental processes. All described canonical configurations of BAF require the presence of a subunit containing an AT-rich DNA binding domain (ARID). Namely, ARID1B and its paralog ARID1A encode for the two largest, mutually exclusive, subunits of the complex: ARID1A and ARID1B. Interestingly, the ARID1B gene, but not ARID1A, is among the most commonly mutated genes in ASD and in syndromic and non-syndromic IDs [59]. Mutations observed include deletions and truncations due to de novo nonsense or frameshift mutations as well as translocations leading to ARID1B haploinsufficiency in the individuals with intellectual disability. Furthermore, de novo haploinsufficient mutations in this gene are found in ~75% of Coffin-Siris syndrome patients, a neurodevelopmental condition characterized by learning disability and craniofacial features. Despite the biochemical knowledge on their mechanisms of action, the roles of chromatin remodeling factors during brain development have not been identified. Furthermore, the brain defects that arise as a consequence of changes in chromatin structure, dynamics and function remain largely unexplored. Paglaroli et al. [60] have studied the molecular role of ARID1B in cell fate commitment during neurodevelopment. Using ARID1B+/– Coffin-Siris patient-derived hiPSCs, they described for the first time that a switching mechanism between ARID1B containing BAF (ARID1B-BAF) and ARID1A-BAF is required for lineage specification and for exiting the pluripotent state. In PSCs, pluripotency is conveyed via binding of an ARID1A-containing BAF to pluripotency-associated enhancers of the SOX2 and NANOG networks. In order to perform transition to a multipotent state, ARID1B-BAF is transiently activated to replace ARID1A-BAF at the SOX2/NANOG enhancers and elicits their repression (see Fig. 2). The haploinsufficient mutation of ARID1B in Coffin-Siris syndrome impairs the switch from ARID1A-BAF to ARID1B-BAF, thus maintaining ARID1A-BAF at pluripotency enhancers. This leads to aberrant binding of SOX2 that interferes with the gene expression patterns required for neurodevelopment. Altogether, these alterations result in an impaired cranial neural crest differentiation that accounts for the phenotypes observed in Coffin-Siris patients. These findings provide the molecular mechanisms underlying not only Coffin-Siris syndrome but also in some non-syndromic IDs and ASD in which ARID1B is frequently mutated [59].
Fig. 2. Schematic representation of ARID1A-BAF switch to ARID1B-BAF during PSC to multipotent state transition (top). In ARID1B+/- mutations, ARID1A-BAF is maintained at pluripotent enhancers, leading to an impaired neurodevelopment (bottom). tRNAs methylation and Intellectual disability Transfer RNAs (tRNA) are small and highly abundant RNA molecules in the cell, representing around 10% of the total cell’s RNA pool [61]. tRNA help decode a messenger RNA (mRNA) sequence into a protein, coupling transcriptional and translational processes. tRNAs are highly structured and contain a high number of post-translational modifications such as methylation and acylation. These modifications contribute to the stability and integrity of tRNAs, to the codon-anticodon interaction, which contributes to translation efficiency and to the RNA quality control and regulation of tRNA localization in the cell. Neuronal function requires high translation efficiency due to the high translation rate and alterations in translation are now considered a pivotal pathogenic mechanism in several neurologic disorders, especially in NDDs [62]. One of the most common tRNA modifications is methylation, with more than 30 methylated nucleotides found at different positions in tRNAs. tRNAs methylation is fundamental for the normal function of different organs, and the disruption of this process is associated with cancer, neurodegeneration and intellectual disability (ID) and neurological phenotypes are often the primary manifestation of mutations affecting the tRNA regulome [63, 64]. tRNA methyltransferase genes such as TRMT10A, TRMT1, FTSJ1, ELP1, WDR4 and NSUN2 have been linked to different forms of ID [65-67]. The human FTSJ1RNA 2’-O-methyltransferase 1 (FTSJ1) gene is located on the X chromosome and several mutations in this gene have been associated with the development of X-linked intellectual disability (XLID) in several families [68]. Nagayoshi et al. have analyzed why the loss of FTSJ1 and its methyltransferase activity would lead to the development of ID despite its ubiquitous expression [69]. Using Ftsj1 KO mice and XLID patient-derived cells, they observed a reduction in 2’O-methylated nucleotides in 11 species of tRNA including tRNAPhe, a Ftsj1 substrate. Among all tRNA species, tRNAPhe is the most reduced in Ftsj1 KO mouse brain compared to the subtle reduction of the other tRNAs. tRNA-sequencing demonstrated an accumulation of fragments of tRNAPhe in the Ftsj1 KO mouse brains. These findings revealed that the absence of the Ftsj1 methylation leads to a cleavage in specific positions, and, ultimately, to perturbed decoding at phenylalanine (Phe) codons. The authors hypothesized that alterations in Phe codon translation leads to dysfunction in the translation of fundamental proteins for neuron function as they found dysregulation in genes involved in synapse organization and cell projection, including Wnt7b and Ctnnb1 as two interesting candidates related to the Wnt signaling pathway [70]. Ftsj1 KO showed an increase of thin dendritic spines indicating immaturity of synapses, and a decrease in the length and width of the postsynaptic density indicating alterations in connectivity. Finally, the authors demonstrate that Ftsj1 KO mice show altered LTP and LTD, and behavioral impairment with increased anxiety, slower spatial learning and poor associative learning. These results establish for the first time how alterations in the normal physiology of tRNA methyltransferases lead to aberrant translation, altered cell biology, impaired electrophysiological properties and, ultimately, behavioral deterioration. Mitovesicles: a new player in the neuropathology of neurodevelopmental disorders Mitochondria, the organelles classically seen as the powerhouse of the cell, also play a role in a plethora of crucial cellular functions, from regulation of calcium homeostasis, initiation of apoptosis, thermoregulation, red-ox balance, to epigenetic regulation by means of methylation, acetylation, or phosphorylation. Genes encoding mitochondrial proteins are increasingly associated with a wide variety of NDDs such as epileptic encephalopathy, ID, or ASD and recent reports show that proteins associated with intellectual disability are linked to mitochondrial function. Interestingly, mitochondrial components can be mediators for intra- and inter-cellular communication under physiological and pathological conditions, being transferred through extracellular vesicles (EVs). The study of EVs, defined as heterogeneous nanoscale vesicles secreted into the extracellular space, is a growing field in biomedicine and particularly in neuroscience due to its potential as a biomarker. Microvesicles and exosomes are the main types of EV, with subtle differences in composition [71]. In a recent study, the ability of EVs to transfer mitochondrial components was examined in Fmr1 knockout mice, a model of FXS [72]. The authors found reduced mitochondrial components in mitochondrial fractions from cortical tissues and astrocytes of Fmr1 KO mouse brains, and were able to monitor mitochondrial dysfunction in EVs. In fact, mitochondrial components are the least studied components in EVs [73]. During years, there was a controversy about the nature of this secreted mitochondrial material with evidences suggesting the secretion of exosomes packed with mitochondrial material or the secretion of whole mitochondria, leading to exchange of these organelles between cells [74-76]. However, the lack of standardized methodologies to accurately separate these different components impeded the proper study of the biological mechanisms behind the different subtypes of EVs [77]. D’Acunzo et al. now developed a new method based on density gradients to achieve high resolution isolation of different EV subpopulations. This new method led to the finding of a new class of previously unknown, mitochondria-derived EVs [78]. First, by using a high-resolution step gradient, the authors purified different EV fractions from mouse brains and demonstrated that the content and composition of EVs was different between the isolated fractions. Using cryogenic electron microscopy, they identified three brain EV subtypes based on protein content, morphology, electron density and size. Then, they analyzed the protein composition of these different fractions, and identified a new class of vesicles with high abundance in mitochondrial markers, “mitovesicles”, of mitochondrial origin. Using liquid chromatography-mass spectrometry (LC-MS) on the fraction enriched with mitovesicles, the authors provide data about their protein composition, with 279 mitochondria-specific proteins of 673 proteins identified in this fraction. Alterations in mitochondria, from the fetal to the adult stage, are a pathological signature of patients with Down syndrome (DS) [79]. The authors hypothesized that the newly described mitovesicles were aberrant in DS. In a trisomic mouse model of DS, Ts[Rb(12.17)]2Cje (Ts2), the authors found alterations in mitochondrial proteins in EV fractions and a higher number of mitovesicles in the parenchyma. They replicated these results in humans using post mortem brains of individuals with DS. Although the pathogenic role of these alterations in number and in protein composition of mitovesicles remains unknown, DS cells are deficient in the elimination of mitochondria, a process called mitophagy, and thus, the excess of mitovesicles could contribute to the removal of damaged mitochondria, working as a homeostatic process to reduce cellular stress. Another possibility is that mitovesicles may activate microglia through acting as proinflammatory agents, which would explain the over-activated microglia detected in several DS mouse models [80]. This study describes and characterizes for the first time a new type of EV derived from mitochondria and also describes a potential role in an NDD such as DS. Discussion The complexity and heterogeneity of the myriad of NDDs makes it difficult to decipher the ultimate mechanisms of NDD neuropathology. Accumulating evidence suggests that their multidimensional nature can be explained by the interaction of numerous genetic and environmental factors. Luckily, we are now facing exciting times in which we can study neurodevelopmental disorder from a holistic perspective using advanced omics and cell engineering techniques that are increasing the temporo-spatial resolution of our mechanistic understanding. Techniques such as single cell omics or spatial transcriptomics and proteomics help us to unravel the enormous diversity and complexity of both cell composition and cell type-specific molecular signatures throughout the brain. The number of publications associating new cellular players such as the astrocytes with brain alterations have increased exponentially in the last three decades suggesting an important role of this cell type in the onset and progression of several NDDs. Every year, selected discoveries open new avenues to investigate NDDs from completely unexplored perspectives and serve as building blocks to better comprehend the enormous complexity of NDDs, hopefully leading to better diagnostics and new therapeutic avenues. Acknowledgements The lab of MD is supported by the Secretaria d’Universitats i Recerca del Departament d’Economia I Coneixement de la Generalitat de Catalunya (Grups consolidats 2017 SGR 926). We also acknowledge the support of the Agencia Estatal de Investigación (PID2019-110755RB-I00/AEI / 10.13039/501100011033), H2020 SC1 GO-DS21-848077 and 101057454-Psych-STRATA, Jerôme Lejeune Foundation #2002, NIH (Grant Number: 1R01EB 028159-01), Fundació La Marató-TV3 (#2016/20-30), Ministerio de Ciencia Innovación y Universidades (RTC2019-007230-1 and RTC2019-007329-1). CS received an FI grant (2020FI_B2 00143) from the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) de la Generalitat de Catalunya; MS received an FPU fellowship (FPU19/04789) and AFB received an FPI-SO fellowship (PRE2018-084504) of the Ministerio de Universidades, Spain. We acknowledge support of the Spanish Ministry of Science and Innovation to the EMBL partnership, the Centro de Excelencia Severo Ochoa and the CERCA Programme /Generalitat de Catalunya. The CIBER of Rare Diseases is an initiative of the ISCIII. References
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |