|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Free Neuropathology 3:1 (2022) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Case Report |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Medulloblastoma and Cowden syndrome: Further evidence of an association |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Steffen Albrecht1,*, Barbara Miedzybrodzki2, Laura Palma3,4, Van Hung Nguyen1, Roy W.R. Dudley5, Torsten Pietsch6, Tobias Goschzik6, Nada Jabado3,7,8, Catherine Goudie8,9, William D. Foulkes3,10,11,12 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
1 Department of Pathology, McGill University, Montreal, QC, Canada |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Corresponding author: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Submitted: 02 December 2021 Accepted: 02 January 2022 Copyedited by: Jeffrey Nirschl Published: 11 January 2022 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Keywords: Medulloblastoma, Cowden syndrome, PTEN |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Abstract Cowden syndrome (CS) is an autosomal dominant hamartoma and tumor predisposition syndrome caused by heterozygous pathogenic germline variants in PTEN in most affected individuals. Major features include macrocrania, multiple facial tricholemmomas, acral and oral keratoses and papillomas, as well as mammary, non-medullary thyroid, renal, and endometrial carcinomas. Lhermitte-Duclos disease (LDD), or dysplastic gangliocytoma of the cerebellum, is the typical brain tumor associated with CS; the lifetime risk for LDD in CS patients has been estimated to be as high as 30%. In contrast, medulloblastoma is much rarer in CS, with only 4 reported cases in the literature. We report a 5th such patient. All 5 patients were diagnosed between 1 and 2 years of age and not all showed the pathognomonic clinical stigmata of CS at the time of their medulloblastoma diagnosis. Where detailed information was available, the medulloblastoma was of the SHH-subtype, in keeping with the observation that in sporadic medulloblastomas, PTEN-alterations are usually encountered in the SHH-subtype. Medulloblastomas can be associated with several tumor-predisposition syndromes and of the 4 medulloblastoma subtypes, SHH-medulloblastomas in children have the highest prevalence of predisposing germline variants (approx. 40%). CS should be added to the list of SHH-medulloblastoma-associated syndromes. Germline analysis of PTEN should be performed in infants with SHH-medulloblastomas, regardless of their clinical phenotype, especially if they do not carry pathogenic germline variants in PTCH1 or SUFU, the most commonly altered predisposing genes in this age-group. In addition, these cases show that CS has a biphasic brain tumor distribution, both in regards to the age of onset and the tumor type: a small number of CS patients develop a medulloblastoma in infancy while many more develop LDD in adulthood. Case history The patient was born at term after an uneventful pregnancy; however, she was noted to have pronounced frontal bossing and macrocrania, (head circumference > 98th percentile). She presented at age 15 months because of rapidly increasing head circumference. MRI showed an extra-axial posterior fossa mass within the cisterna magna, measuring 5.3 x 4.4 x 3.2 cm. The tumor was resected; post-operative MRI confirmed gross total resection. Histologically, it was a medulloblastoma with extensive nodularity (MBEN). She was treated according to the CCG-99703 protocol, which consists of 3 cycles of induction chemotherapy followed by 3 cycles of marrow-ablative consolidation chemotherapy with autologous stem-cell rescue [Cohen et al. 2015]; however, she only received 2 of the latter due to hematological toxicity. During follow-up, some keratotic papules on the fingers and buttocks were noted and therefore Gorlin syndrome (nevoid basal cell carcinoma syndrome; Online Mendelian Inheritance in Man (OMIM) #109400) was suspected clinically. Some of these cutaneous lesions were biopsied but consisted of non-specific keratoses or warts; there were no basal carcinomas. Initial germline genetic testing at age 2 consisted of deletion analysis and Sanger sequencing of PTCH1, which were negative. Sanger sequencing of SUFU was done at age 5 and was negative. At age 8, a firm papule was noted on the neck; histologically, it was a sclerotic fibroma. (A similar lesion was also noted on the left thigh, but not excised.) At age 9, four palmar pits were noted on the right hand. Although the combination of palmar pits, MBEN, and macrocrania was suggestive of Gorlin syndrome, sclerotic fibroma has been associated with Cowden syndrome (CS; OMIM #158350) [Kieselova et al. 2017]. Therefore, next-generation germline sequencing of PTCH1, PTCH2, SUFU, and PTEN was performed. A previously reported pathogenic variant in PTEN, c.388C>T, p.R130* (COSV64288463), was identified. No variants were found in the other genes. Parental studies were negative, suggesting most likely a de novo mutation, although gonadal mosaicism in one of the parents cannot be ruled out. At age 10, a posterior fossa dural arterio-venous fistula (DAVF) was suspected on a routine follow-up MRI and confirmed by cerebral angiogram to be a Borden type 1 DAVF draining into an ectopic pouch of the right transverse sinus; it was fed by branches of the right and left external carotid arteries and the right posterior cerebral artery. Although hemorrhage from intracranial AVFs has been reported in patients with CS [Prats-Sánchez et al. 2016], Borden type 1 DAVFs have a very low risk of hemorrhage [Gandhi et al. 2012] and the lesion was completely asymptomatic. It was therefore not treated. It was no longer seen on repeat imaging a year later, suggesting spontaneous involution-resolution. At age 12, multiple thyroid nodules were noted bilaterally on ultrasound; fine needle aspiration cytology was consistent with benign follicular nodules. Now age 13 (i.e., 12 years post-diagnosis), the patient is alive and well without evidence of recurrent medulloblastoma. She does not have facial or oral lesions.
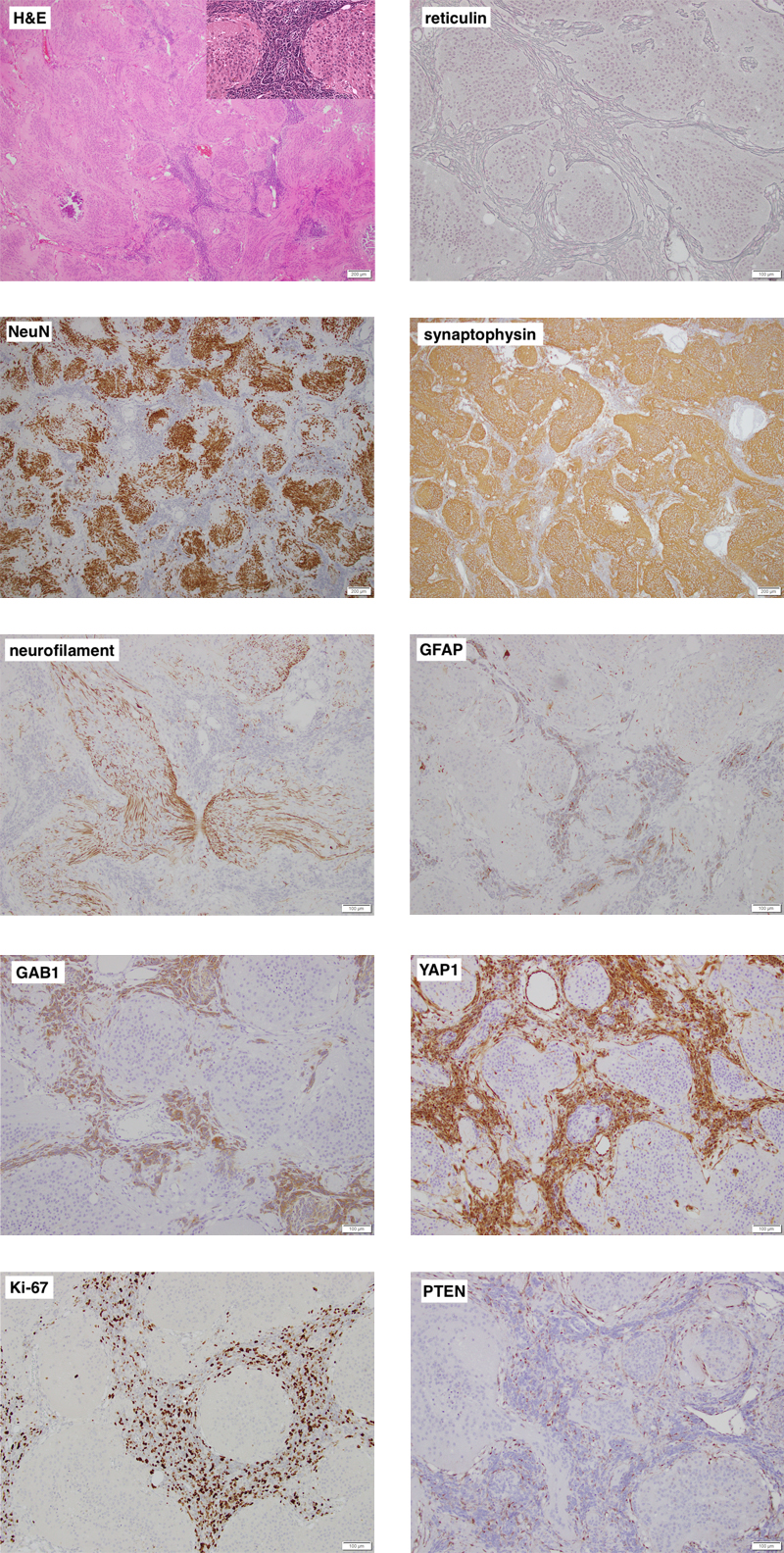
Figure 1. Representative images of the cerebellar tumor’s histology and immunophenotype. Individual images are labelled with the respective stain. On low power, the tumor is biphasic and distinctly nodular. Most of the tumor is made up of neurocytic to gangliocytic cells (high power inset) that are strongly NeuN immunoreactive and embedded in a synaptophysin-positive neuropil-like matrix that contains bundles of neurites. The highly cellular desmoplastic component shows some staining for GFAP and is strongly positive for GAB1 and YAP1. Ki-67 labelling is seen almost exclusively in the desmoplastic component. Both components lack expression of PTEN, which is retained in endothelial cells. Clicking the figure will lead you to the full virtual slides. Pathology Representative images of the cerebellar tumor are shown in Figure 1. The tumor was biphasic and had a distinctly nodular growth pattern. Most of the tumor consisted of large confluent islands of isomorphic cells with a neurocytic to gangliocytic appearance, embedded in a finely fibrillary neuropil-like matrix. These cells were strongly positive for NeuN and the neuropil stained intensely for synaptophysin and showed bundles of neurites on a neurofilament immunostain. There was virtually no Ki-67 labelling in this component and it was negative for YAP1 and GAB1. The remainder of the tumor was made up of a very cellular "small blue cell" component, which was rich in reticulin fibers, had a high mitotic rate with approximately 50% Ki-67 labelling, and focal immunoreactivity for GFAP along with strong staining for YAP1 and GAB1. The features were typical of an MBEN, SHH-activated. Immunostaining for PTEN and genetic analysis of the tumor were performed after the PTEN germline variant was identified. Expression of PTEN was retained in vessels but lost in both tumor components. The tumor carried the PTEN p.R130* variant, as expected. In addition, using a TruSeq custom amplicon (Illumina) for 13 genes frequently mutated in SHH-medulloblastomas [Goschzik et al. 2021], a known pathogenic variant in SMO, c.1234C>T, p.L412F (COSV50824425), was identified, which was confirmed by Sanger sequencing. Molecular inversion probe technology was used to perform high-resolution genome-wide copy number analysis as described previously [Wang et al. 2012]. It showed a stable genome with copy-neutral allelic loss on chromosome arm 10q. There was no amplification of MYC or MYCN. Discussion Cowden syndrome (OMIM #158350) is an autosomal dominant hamartoma and tumor predisposition syndrome caused by heterozygous pathogenic germline variants in PTEN in about 80% of affected individuals [Yehia & Eng 2001/2021]. Major features include macrocrania, multiple facial tricholemmomas, acral and oral keratoses and papillomas, as well as mammary, non-medullary thyroid, renal, and endometrial carcinomas [Yehia & Eng 2001/2021]. Lhermitte-Duclos disease (LDD), or dysplastic gangliocytoma of the cerebellum, is the typical brain tumor associated with CS; in fact, in adults, LDD is considered pathognomonic of CS [Yehia & Eng 2001/2021]. Conversely, the lifetime risk for LDD in CS patients has been reported to be as high as 30% [Riegert-Johnson et al. 2010]. In contrast, the association between CS and medulloblastoma is very rare. In a study of 914 children and adolescents with cancer, including 227 with medulloblastoma, no PTEN germline variants were identified [Gröbner et al. 2018]. Similarly, among 1022 medulloblastoma patients screened for germline variants in 110 cancer predisposition genes, only one pathogenic variant was reported in PTEN [Waszak et al. 2018]. In a clinical cohort of 368 patients with Cowden syndrome and pathogenic PTEN germline variants (including 98 patients under the age of 18 years) recruited and followed prospectively by an international consortium between 2000 and 2010, no cases of medulloblastoma were reported [Tan et al. 2012]. On the other hand, including this case, five patients with CS and medulloblastoma have now been reported in the literature; they are summarized in Table 1. Patient 1 was reported before the genetic defect in CS was known [Bagan et al. 1989], hence the diagnosis of CS is based on the clinical findings [Eng 2000]; he has since been lost to follow-up (J.V. Bagan, personal communication). The four other patients carried a pathogenic germline variant in PTEN. Given the rarity of CS, with an estimated prevalence of 1 in 200,000 [Yehia & Eng 2001/2021], this is unlikely to be a coincidence. Table 1: Patients with Cowden syndrome and medulloblastoma.
Abbreviations: DN, desmoplastic nodular; MB, medulloblastoma; MBEN, medulloblastoma with extensive nodularity; mo, month; na, not applicable; nd, not done; NED, no evidence of disease; ns, not stated; y, year ¶ R. Niinimäki, personal communication Interestingly, in the three cases in which a detailed analysis of the medulloblastoma was performed, it was an SHH-activated medulloblastoma. Two of them also had an additional pathogenic variant in a component of the SHH-pathway and 2 showed allelic loss involving PTEN/10q. This mirrors findings in sporadic medulloblastomas, where mutations or deletions of PTEN are more frequent in SHH-medulloblastomas than in other medulloblastoma subtypes [Northcott et al. 2012]. Furthermore, in mouse models of SHH-medulloblastomas driven either by inactivation of Ptch1 [Metcalfe et al. 2013] or activation of Smo [Castellino et al. 2010], additional loss of Pten changed the tumor histology from tumors resembling classic medulloblastomas to tumors resembling MBEN. Using similarity network fusion applied to genome-wide DNA methylation and gene expression data, SHH-medulloblastomas can be divided into 4 distinct subtypes [Cavalli et al. 2017]; we did not perform this type of analysis on our patient’s tumor and based on the published data, neither did the authors of the two other cases where the tumor was analyzed (cases 3 and 4). Brain-specific suppression of Pten in mice produces cerebellar lesions resembling LDD, but not medulloblastomas [Backman et al. 2001; Kwon et al. 2001]. In human SHH-medulloblastomas, loss of PTEN almost never occurs in isolation. In a detailed analysis of 196 sporadic SHH-medulloblastomas [Skowron et al. 2021, figure 2A], nine had a deletion and/or mutation of PTEN. Eight of these tumors had additional, often multiple, genetic alterations, typically involving a gene of the SHH-pathway, loss of 9q (which deletes PTCH1), loss of 10q (which deletes PTEN and SUFU), or mutations and fusions in other genes. Only one tumor had an isolated variant in PTEN, but the analyses may not have captured all possible genetic alterations (P. Skowron and M. Taylor, personal communications). These findings indicate that in most instances, loss of PTEN alone is insufficient to produce medulloblastomas. SHH-activated medulloblastomas are thought to arise from granule cell precursors that form the external granular layer (EGL) of the cerebellar cortex; their proliferation and inward migration to form the internal granule cell layer is driven by Purkinje cell-derived SHH [Tamayo-Orreo & Charron 2019]. In humans, the EGL persists after birth. It starts to involute by the 2nd to 4th post-natal month and usually has disappeared by 1 year, although rare cases of persistence until 2 years have been reported [Friede 1989]. In a patient with CS, all granule cell precursors in the EGL are haploinsufficient for PTEN, but this is not sufficient to produce a medulloblastoma. However, if one of these cells acquires additional alterations of the type described by Skowron et al. [Skowron et al. 2021], it can give rise to an SHH-medulloblastoma. This would be a rare event, resulting in a low frequency of medulloblastomas in CS patients. Furthermore, given the rapid disappearance of the EGL after birth, the "window of opportunity" for the development of a medulloblastoma in CS is brief and explains why they only occur in infants in these patients. CS may be under-recognized in medulloblastoma patients. While 90% of patients with CS will have some features of CS by age 20 [Yehia & Eng 2002/2021], infants and young children may not show some of the pathognomonic features, such as multiple facial tricholemmomas, and present instead with non-specific findings, like macrocrania, autism, or developmental delay [Busa et al. 2015]. Among the 5 patients with CS and medulloblastoma, patient 1 had oral papillomas at the time of his medulloblastoma diagnosis [Bagan et al. 1989]. Patient 2 developed gastro-intestinal polyps after his medulloblastoma and sought medical attention for oral and cutaneous lesions at age 14, at which time the diagnosis of CS was made [Palatini et al. 2016]. No clinical details are available on patient 3 [Waszak et al. 2018]. Patient 4 showed only macrocrania at the time her medulloblastoma was diagnosed [Tolonen et al. 2020] and still does not have other stigmata of CS (R. Niinimäki, personal communication). In our patient, sequencing of PTEN was only undertaken at age 9, after the diagnosis of the sclerotic fibroma. Thus, in some of these young patients, the diagnosis of CS was not clinically obvious at the time they presented with a medulloblastoma; we therefore suspect that CS may be underdiagnosed in medulloblastoma patients. A high prevalence of tumor predisposition syndromes in children with SHH-medulloblastomas, especially infants, has long been known. For instance, in a single-institution series of 82 medulloblastoma patients, 6 of 12 patients with an MBEN had a genetic syndrome (5 Gorlin, 1 fragile-X); 5 of these 6 were less than 24 months of age [Garrè et al. 2009]. In the aforementioned series of 1022 medulloblastomas, predisposing germline variants in APC, BRCA2, PALPB2, PTCH1, SUFU, and TP53 occurred in 6% of patients overall, and in 20% of those with SHH-medulloblastomas [Waszak et al. 2018]. A further analysis by the same group [Waszak et al. 2020] identified in addition pathogenic germline variants in ELP1 in 14% of pediatric patients with SHH-medulloblastomas; all in all, 77 of 202 children (38%) with an SHH-medulloblastoma had a predisposing germline variant in one of 7 genes. Here, we present evidence that CS, too, can be associated with SHH-medulloblastomas in infants. Therefore, PTEN should be analyzed in these patients, regardless of their clinical phenotype, especially if they do not carry pathogenic germline variants in PTCH1 or SUFU, the most commonly altered predisposing genes in infant SHH-medulloblastomas [Waszak et al. 2020]. Whether CS also predisposes to other types of medulloblastoma (i.e., WNT, group 3, or group 4) remains to be seen. In addition, these cases show that Cowden syndrome has a biphasic brain tumor distribution, both in regards to the age of onset and the tumor type: a small number of Cowden patients develop a medulloblastoma in infancy while many more develop LDD in adulthood. Acknowledgements We thank J.V. Bagan, R. Niinimäki, S. Pfister, P. Skowron, and M.D. Taylor for kindly providing additional information on previously published cases. W. Foulkes receives funding from the Canadian Institutes of Health Research (grant FDN-148390) and so does N. Jabado (grant MOP-286756 and FDN-154307). References Backman SA et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet 29: 396–403, 2001. https://doi.org/10.1038/ng782 Bagan JV et al. Cowden syndrome: clinical and pathological consideration in two new cases. J Oral Maxillofac Surg 47: 291–4, 1989. https://doi.org/10.1016/0278-2391(89)90234-6 Busa T et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome. Eur J Paediatr Neurol 19: 188–92, 2015. https://doi.org/10.1016/j.ejpn.2014.11.012 Castellino RC et al. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS One 5: e10849, 2010. https://doi.org/10.1371/journal.pone.0010849 Cavalli FMG et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31: 737–54.e6, 2017. https://doi.org/10.1016/j.ccell.2017.05.005 Cohen BH et al. Pilot study of intensive chemotherapy with peripheral hematopoietic cell support for children less than 3 years of age with malignant brain tumors, the CCG-99703 Phase I/II Study. A report from the Children's Oncology Group. Pediatr Neurol 53: 31–46, 2015. https://doi.org/10.1016/j.pediatrneurol.2015.03.019 Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 37: 828–30, 2000. https://doi.org/10.1136/jmg.37.11.828 Friede RL. Developmental Neuropathology. 2nd ed, Springer, 1989, p. 15–6. Gandhi D et al. Intracranial dural arteriovenous fistulas: classification, imaging findings, and treatment. AJNR Am J Neuroradiol. 33: 1007–13, 2012. https://doi.org/10.3174/ajnr.A2798 Garrè ML et al. Medulloblastoma variants: age-dependent occurrence and relation to Gorlin syndrome—a new clinical perspective. Clin Cancer Res 15: 2463–71, 2009. https://doi.org/10.1158/1078-0432.CCR-08-2023 Goschzik T et al. Medulloblastoma in adults: cytogenetic phenotypes identify prognostic subgroups. J Neuropathol Exp Neurol 80: 419–30, 2021. https://doi.org/10.1093/jnen/nlab020 Gröbner SN et al. The landscape of genomic alterations across childhood cancers. Nature 555: 321–27, 2018. https://doi.org/10.1038/nature25480 Kieselova K et al. Multiple sclerotic fibromas of the skin: an important clue for the diagnosis of Cowden syndrome. BMJ Case Rep 2017. https://doi.org/10.1136/bcr-2017-221695 Kwon CH et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 29: 404–11, 2001. https://doi.org/10.1038/ng781 Metcalfe C et al. PTEN loss mitigates the response of medulloblastoma to Hedgehog pathway inhibition. Cancer Res 73: 7034–42, 2013. https://doi.org/10.1158/0008-5472.CAN-13-1222 Northcott PA et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488: 49–56, 2012. https://doi.org/10.1038/nature11327 Prats-Sánchez LA et al. Multiple intracranial arteriovenous fistulas in Cowden syndrome. J Stroke Cerebrovasc Dis 25: e93–4, 2016. https://doi.org/10.1016/j.jstrokecerebrovasdis.2016.03.048 Riegert-Johnson DL et al. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract 8: 6, 2010. https://doi.org/10.1186/1897-4287-8-6 Skowron P et al. The transcriptional landscape of Shh medulloblastoma. Nat Commun 12: 1749, 2021. https://doi.org/10.1038/s41467-021-21883-0 Tamayo-Orrego L, Charron F. Recent advances in SHH medulloblastoma progression: tumor suppressor mechanisms and the tumor microenvironment. F1000Res 8: F1000 Faculty Rev-1823, 2019. https://doi.org/10.12688/f1000research.20013.1 Tan MH et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18: 400–7, 2012. https://doi.org/10.1158/1078-0432.CCR-11-2283 Tolonen JP et al. Medulloblastoma, macrocephaly, and a pathogenic germline PTEN variant: cause or coincidence? Molec Genet Genomic Med 8: e1302, 2020. https://doi.org/10.1002/mgg3.1302 Waszak SM et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet. Oncol 19: 785–98, 2018. https://doi.org/10.1016/S1470-2045(18)30242-0 Waszak SM et al. Germline Elongator mutations in Sonic Hedgehog medulloblastoma. Nature 580: 396–401, 2020. https://doi.org/10.1038/s41586-020-2164-5 Yehia L, Eng C. PTEN Hamartoma Tumor Syndrome. 2001 Nov 29 [Updated 2021 Feb 11]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021.
Copyright: © 2022 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |