|
|
|
Free Neuropathology 2:3 (2021) |
|
Review |
|
Neuromuscular disease: 2021 update |
|
Marta Margeta |
|
Department of Pathology, University of California, San Francisco, CA, USA |
|
Corresponding author: |
|
Submitted: 5 February 2021 Accepted: 22 February 2021 Copyedited by: Henry Robbert Published: 23 February 2021 |
|
Keywords: Costameres, Sarcomeres, Satellite cell activation, COVID-19, Guillain-Barré syndrome, 5q-SMA, SMALED, Lobulated fibers, Supervillin, Vacuolar myopathy, Perilipin 4, Sarcolemmal repair, TRIM72, Vasculitis, Amyloidosis, Gene therapy, Large-scale electron microscopy |
|
Abstract This review highlights ten important advances in the neuromuscular disease field that were first reported in 2020. The overarching topics include (i) advances in understanding of fundamental neuromuscular biology; (ii) new / emerging diseases; (iii) advances in understanding of disease etiology and pathogenesis; (iv) diagnostic advances; and (v) therapeutic advances. Within this broad framework, the individual disease entities that are discussed in more detail include neuromuscular complications of COVID-19, supervillin-deficient myopathy, 19p13.3-linked distal myopathy, vasculitic neuropathy due to eosinophilic granulomatosis with polyangiitis, spinal muscular atrophy, idiopathic inflammatory myopathies, and transthyretin neuropathy/myopathy. In addition, the review highlights several other advances (such as the revised view of the myofibrillar architecture, new insights into molecular and cellular mechanisms of muscle regeneration, and development of new electron microscopy tools) that will likely have a significant impact on the overall neuromuscular disease field going forward. Abbreviations AAV, adeno-associated virus; ANCA, antineutrophil cytoplasmic antibody; ASM, antisynthetase syndrome-associated myositis; ATTR, transthyretin amyloidosis; CGRP, calcitonin-gene related peptide; COVID-19, Coronavirus Disease 2019; CK, creatine kinase; DM, dermatomyositis; FESEM, field-emission scanning electron microscopy; GLUD1, glutamate dehydrogenase 1; EGPA, eosinophilic granulomatosis with polyangiitis; GBS, Guillain-Barré syndrome(s); HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; IIM, idiopathic inflammatory myopathy; IMNM, immune-mediated necrotizing myopathy; LMN, lower motor neuron; MmSc, muscle mesenchymal stromal cells; NMD, neuromuscular disease; SARS, severe acute respiratory syndrome; sIBM, sporadic inclusion body myositis; SMA, spinal muscular atrophy; SMALED, spinal muscular atrophy lower extremity dominant; SRP, signal recognition particle; STEM, scanning transmission electron microscopy; SVV, small vessel vasculitis; TEM, transmission electron microscopy; Tregs, regulatory T cells; TTR, transthyretin.
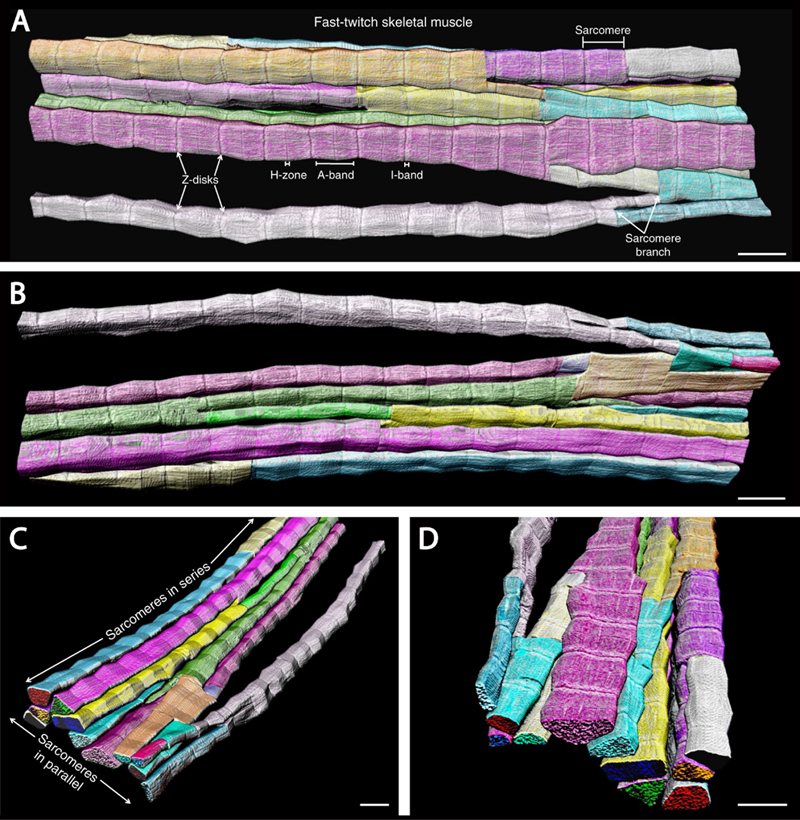
In this update, I will briefly describe ten 2020 neuromuscular field advances that I consider to be most important and/or interesting; as in the prior update (Margeta, 2020b), these advances will be grouped into several different “discovery clusters” and listed in no particular order. Advances in fundamental neuromuscular biology with implications for neuromuscular disease 1. New understanding of the myofibrillar architecture Based on numerous studies that used traditional electron microcopy methods for evaluation of skeletal muscle ultrastructure, the myofibrillar apparatus has long been understood as consisting of independent tube-like myofibrils that are arranged in parallel and span the entire length of a fiber, with each myofibril composed of sarcomeres that are serially connected through Z-discs. This traditional model of the myofibrillar apparatus was effective in explaining how the contractile forces are transmitted down the length of the fiber towards the myotendinous junction, but could not readily explain how these forces were transmitted laterally, towards the sarcolemma and the extracellular matrix (into which myofibers are anchored through costameres, the sarcolemma-associated supramolecular structures that form membrane skeleton and mediate fiber adhesion). A recent groundbreaking study by Willingham et al. has used a new method (focused ion beam-scanning electron microscopy) to image muscle fibers in three dimensions at high resolution, providing a new view of the myofibrillar architecture (Willingham et al., 2020). To their surprise, the authors found that sarcomeres are not organized into independent parallel myofibrils as traditionally thought, but instead form the unified, non-linear myofibrillar matrix that spans both the length and the width of a fiber and is interconnected through extensive and bidirectional sarcomere branching (Fig. 1). This branching occurs in what appears to be a random pattern, with branch points that are not restricted to any specific region of the sarcomere; however, the frequency of branches is fiber-type specific, with branches approximately three times more frequent in slow twitch than fast twitch fibers (and approximately two times more frequent in slow twitch fibers than cardiac myocytes). Interestingly, sarcomere branching is developmentally regulated: while present at birth, it is downregulated in the period of rapid postnatal muscle growth and then upregulated again late in development. Three different mechanisms of sarcomere branching were identified: sarcomere splitting (where some of the myofilaments from one sarcomere separated from the rest, forming two distinct myofibrillar segments), myofilament transfer (where some of the myofilaments were redistributed between sarcomeres, separating from one myofibrillar segment and joining the adjacent one) and myofilament trade (where myofilament transfer between two adjacent myofibrillar segments was reciprocal/bilateral). While most of the work was performed on murine samples, the few human samples that were evaluated in this study showed essentially identical findings, except for a slightly higher branching frequency in human compared to murine fast-twitch fibers (2.4 ± 0.2 vs. 1.6 ± 0.2 branches per 10 sarcomeres); however, a more thorough investigation of inter-species differences remains to be done.
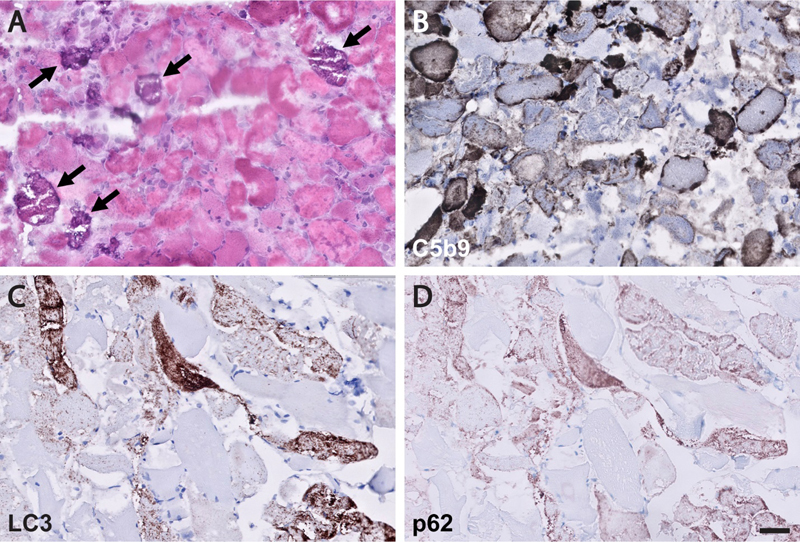
Figure 1. 3D renderings of the interconnected sarcomeres that form the unified myofibrillar matrix. A. 3D rendering of 115 directly connected sarcomeres within the myofibrillar matrix of a fast-twitch muscle. Individual colors represent different myofibrillar segments linked by branching sarcomeres. B-D. 3D renderings at different perspectives highlight the nonlinearity and connections between myofibrillar segments. Representative of 7,707 sarcomeres from four volumes from four mice. Scale bars, 2 µm. (This figure and its legend were adopted from Figure 1 in Willingham et al., 2020; this use is permitted under the Creative Commons Attribution 4.0 International License.) What is the relevance of this new understanding of the myofibrillar architecture for neuromuscular disease (NMD)? That must be experimentally addressed and therefore remains to be seen, but it is very likely that abnormal branching of sarcomeres contributes to poor contractile force generation in various congenital / structural myopathies, which show different degrees of organelle malpositioning and myofilament disorganization. In addition, this new discovery may have important implications for pathogenesis of muscular dystrophies: given that the myofibrillar matrix is directly connected to the costameric membrane skeleton (discussed further in advance #4 below), it is possible that disruption of these connections may result in inappropriate transfer of lateral mechanical forces from the interior of the fiber to the extracellular matrix, thereby contributing to the likelihood of segmental fiber necrosis that over time leads to exhaustion of the muscle regenerative capacity and muscle wasting. 2. Deepening insights into molecular and cellular mechanisms of muscle regeneration Over the past decade, much has been learned about the mechanisms of skeletal muscle regeneration; the key role of satellite cells (muscle stem cells) in this process was first demonstrated in 2011, and the work that followed since then has showed that satellite cell activation requires cytokines and growth factors released by FoxP3-expressing regulatory T cells (Tregs) and macrophages. Two studies published last year build further on these discoveries by elucidating the intercellular crosstalk and chemical signals that mediate muscle regenerative response. The first study focused on the interplay between intramuscular peripheral nerve twigs, muscle mesenchymal stromal cells (MmSCs), and Tregs in skeletal muscle regeneration (Wang et al., 2020). Following muscle injury, MmSCs are the main source of interleukin (IL)-33, which drives subsequent accumulation of Tregs required for satellite cell activation and muscle repair. Wang et al. have found that IL-33+ MmSCs are located in close proximity to peripheral nerve bundles and that they titrate their Il-33 production in response to calcitonin-gene related peptide (CGRP), which plays a role in pain perception and is released by sensory peripheral nerve terminals in response to tissue damage. In agreement with these findings, systemic administration of CGRP led to an increase in the muscle IL-33 level and recruitment of Tregs into the muscle compartment. However, it remains to be shown whether CGRP treatment will enhance muscle regeneration and – conversely – whether inhibition of CGRP signaling will attenuate effective repair following muscle injury; the latter question is particularly pressing given that anti-CGRP antibodies and CGRP antagonists have recently been approved for prevention of migraine headaches. In addition, it remains to be seen whether effective muscle repair requires only signaling from sensory nerve terminals, or whether motor nerve twigs also play a role in this process. The second and more comprehensive of the two studies centered on the nature of chemical crosstalk between macrophages and satellite cells that is required for muscle repair (Shang et al., 2020). The authors of that study modulated glutamine secretion from macrophages via genetic or pharmacologic manipulation of glutamine synthetase (which generates glutamine by condensation of glutamate and ammonia) or glutamate dehydrogenase 1 (GLUD1, which catalyzes oxidative deamination of glutamate and thereby reduces the amount of glutamate available for glutamine synthesis); in addition, they blocked satellite cell glutamine uptake by knocking down glutamine transporter SLC1A5 selectively in those cells. Using these experimental approaches both in vitro and in vivo, the authors demonstrated that satellite cells take up glutamine that is secreted by monocyte-derived macrophages recruited into skeletal muscle following injury; this glutamate uptake, which leads to activation of mTOR signaling, is required for satellite cell proliferation and differentiation. Excitingly, Shang et al. showed that increased glutamine bioavailability in injured or aged muscle translates into increased muscle regenerative capacity: mice with decreased GLUD1 activity showed faster and more effective fiber regeneration following acute myotoxic injury when young, as well as increased muscle mass and improved baseline physical performance when aged. While these findings need to be replicated in other model systems and ultimately human clinical trials, they hold promise not only for treatment of muscular dystrophies and other skeletal myopathies characterized by recurring muscle fiber necrosis, but also for treatment of the age-associated sarcopenia that leads to significant morbidity and mortality in elderly populations. Newly defined / emerging neuromuscular diseases 3. Neuromuscular complications of COVID-19 2020 has been dramatically shaped by the COVID-19 pandemic, which has affected the NMD field in addition to all other aspects of life. COVID-19 is caused by infection with SARS-CoV-2, an RNA virus from the coronavirus family; the disease is asymptomatic in up to 50% of patients, but can also cause severe pneumonia, acute respiratory distress syndrome, and death. Neurologic complications of COVID-19 were noted very early in the pandemic (Mao et al., 2020) and include CNS, PNS, and skeletal muscle manifestations (Cagnazzo et al., 2020); in this update, two forms of potential neuromuscular involvement [Guillain-Barré syndromes (GBS) and rhabdomyolysis / myopathy] will be discussed in more detail. GBS are a group of acute immune-mediated polyneuropathies that are characterized by ascending weakness, mild-moderate sensory abnormalities, and pain; the most common subtypes include acute demyelinating inflammatory polyradiculoneuropathy, acute motor axonal neuropathy, and the Miller-Fischer syndrome (Dalakas, 2020). In approximately 70% of GBS patients, a flu-like illness precedes neurologic symptoms by ~1-3 weeks, and all subtypes of GBS show association with different infectious agents (both viruses and bacteria). Perhaps not surprisingly, GBS was also documented in patients with COVID-19, first through case reports and small case series (reviewed in Caress et al., 2020; Dalakas, 2020; De Sanctis et al., 2020) and later through small retrospective case-control studies, one from Italy (Filosto et al., 2020) and another from Spain (Fragiel et al., 2020). Based on these early reports, COVID-19-associated GBS was shown to have typical clinical characteristics, with most cases falling within the acute demyelinating subtype and showing onset of neurologic symptoms a median of 10-23 days after the COVID-19 diagnosis. The two retrospective case-control studies found 3- to 10-fold higher GBS incidence during the study period (the 2-month pandemic peak in each country) compared to the equivalent period in 2019; however, the conclusions were limited by relatively small sample sizes and a high potential for confounding due to selection and ascertainment bias. Indeed, the most comprehensive investigation of the potential link between GBS and COVID-19 to date (a combined retrospective epidemiologic and prospective cohort study performed in the UK and published at the end of 2020) found no association between these two conditions (Keddie et al., 2020). In fact, and somewhat counterintuitively, the authors of that study found a decrease (rather than increase) in the number of GBS cases that required IVIg treatment during the 2-month pandemic peak compared to prior years; while reasons for this decrease are not entirely clear, one possible explanation is a decrease in the number of infections with known GBS-triggering pathogens as a result of drastic public health actions that were undertaken to control the pandemic. In addition to a lack of increase in GBS incidence, Keddie et al. found no temporal or spatial association between COVID-19 and GBS cases in the UK during the study period, further weakening the likelihood of true association between these two diseases. Given these contradictory findings, additional studies should be done; however, the preponderance of current evidence suggests that the SARS-CoV-2 infection is not a major trigger of autoimmune polyneuropathy. What about skeletal muscle involvement in COVID-19? As with other flu-like illnesses, myalgias are a common symptom of this viral disease; however, actual skeletal muscle injury [as evidenced by elevated creatine kinase (CK) levels] is seen only in a subset (~10-30%) of patients (Dalakas, 2020; Manzano et al., 2020). In some of these “COVID-19 myopathy” cases, muscle injury is very severe, with CK levels greater than 10,000 U/L (Buckholz et al., 2020; Dalakas, 2020; and a number of individual case reports); the most severe case was documented in a patient on rosuvastatin therapy who was infected with SARS-CoV-2 and developed fatal rhabdomyolysis, with the CK level of ~1,000,000 U/L (Anklesaria et al., 2020). However, very few muscle biopsies or autopsy muscle samples from COVID-19 patients have been comprehensively evaluated to date; as a result, very little is known about the mechanisms that underlie COVID 19-associated muscle injury. Skeletal muscle expresses angiotensin-converting enzyme 2, which serves as the cellular receptor for SARS-CoV-2 (Ferrandi et al., 2020); thus, it is conceivable that this virus can invade and directly injure muscle fibers. However, limited studies published thus far have not provided evidence for viral presence in the injured muscle (Manzano et al., 2020; Rosato et al., 2020; Zhang et al., 2020). [One case report noted the presence of coronavirus-like particles in a postmortem muscle sample from a patient who died from COVID-19 but had no symptoms of skeletal muscle injury (Hooper et al., 2020); however, ultrastructural detection of coronaviruses can be challenging (Dittmayer et al., 2020a) and the virus-like particles shown in that paper more closely resemble clathrin-coated vesicles or parts of the rough endoplasmic reticulum than true coronaviruses.] Instead, the currently available data suggest that COVID 19-associated muscle injury is most likely para-infectious / immune-mediated, and can manifest as dermatomyositis-like “type I interferonopathy” (Manzano et al., 2020), myositis (Zhang et al., 2020), or immune-mediated necrotizing myopathy (Fig. 2). This is in agreement with the work done during the SARS epidemic in early 2000s, which suggested that vasculitis or immune-mediated mechanisms, rather than direct viral infection, were a cause of SARS-associated myopathy (Ding et al., 2003; Leung et al., 2005). Given that the available studies are very limited in scope, much additional work is needed to fully elucidate the mechanisms of skeletal muscle injury in COVID-19. However, an intriguing possibility is that both mechanisms play a role, as recently shown for the chikungunya infection-associated myopathy: in that emerging viral disease, muscle pathology is due to activation of the host immune response but is triggered by viral replication in muscle fibers (Lentscher et al., 2020).
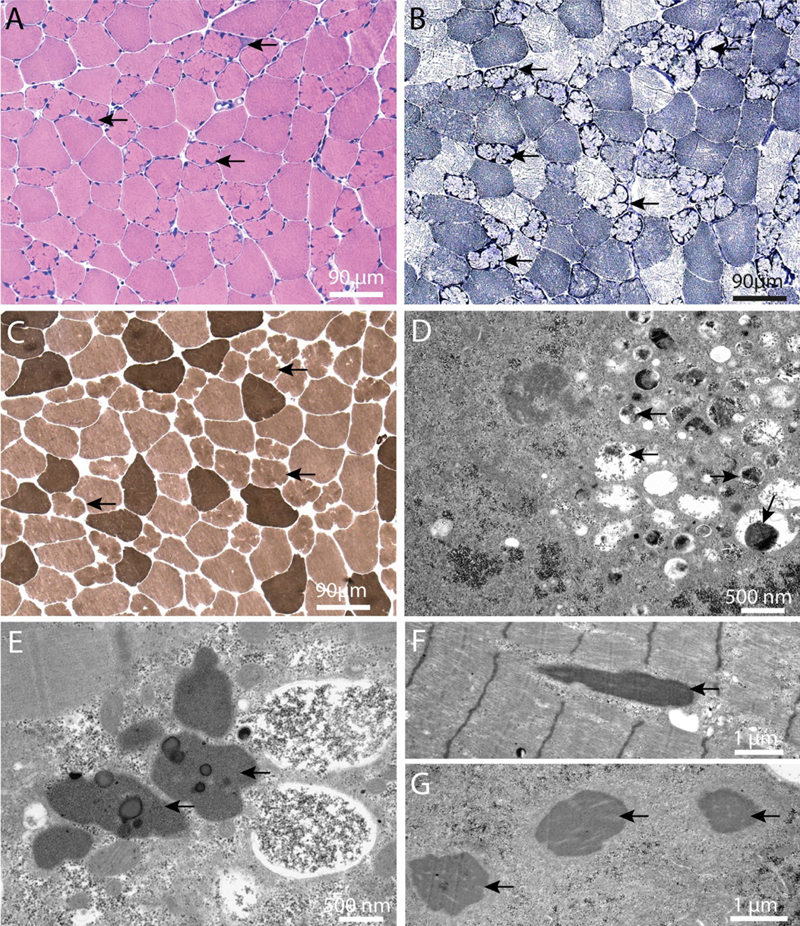
Figure 2. Immune-mediated necrotizing myopathy in a patient with COVID-19. A representative H&E-stained cryosection (A) shows frequent, randomly distributed degenerating/regenerating muscle fibers and focal myophagocytosis in the absence of a significant lymphocytic inflammatory infiltrate; unusual dystrophic calcifications are seen in a subset of necrotic fibers (arrows), confirmed by von Kossa calcium stain (not shown). Deposition of the complement membrane attack complex (C5b9-immunostained cryosection; B) is seen in the sarcoplasm of necrotic fibers as well as the sarcolemma of many intact-appearing fibers. Immunohistochemical stains for LC3 (C) and p62 (D; both performed on formalin-fixed, paraffin embedded tissue) show frequent fibers with densely packed fine puncta. Immunohistochemical stain for SARS-CoV-2 was negative and no coronavirus particles were detected on ultrastructural evaluation (not shown). The patient had a history of hepatitis C, diabetes mellitus, hypertension, and hyperlipidemia treated with atorvastatin. He was diagnosed with COVID-19 based on a positive SARS-CoV-2 PCR test performed following a close exposure; he was asymptomatic at the time, but subsequently developed progressive lower extremity weakness and dysphagia. Approximately 1 month after the first weakness symptoms, he lost his sense of smell, developed shortness of breath, and was hospitalized. At admission, his CK level was 33,000 U/L and he was still positive for SARS-CoV-2; muscle biopsy was performed 10 days into his hospital course (~ 2 months following initial COVID-19 diagnosis). The patient was ultimately treated with prednisone, which led to improvement of his neuromuscular symptoms; he was discharged from the hospital never requiring mechanical ventilation for his COVID-19 pneumonia. Testing for anti-HMGCR and anti-SRP antibodies was not performed. Scale bar, 50 µm. 4. Structural myopathy caused by supervillin deficiency The presence of occasional lobulated fibers is a relatively common and therefore nonspecific finding in muscle biopsies. In some cases, however, frequent lobulated fibers dominate the overall histopathologic picture, resulting in a descriptive diagnosis of a “lobular” (or “trabecular”) myopathy. In general, this descriptive diagnosis does not correspond to a specific, well-defined clinicopathologic entity; however, a newly defined structural myopathy caused by loss-of-function mutations in supervillin (Hedberg-Oldfors et al., 2020) is an exciting exception to this rule and, while probably rare, should be on the radar screen of every neuromuscular pathologist going forward. Supervillin is a large, differentially spliced and ubiquitously expressed actin-binding protein that is particularly abundant in striated muscle; the 250 kDa muscle isoform (also called archvillin) interacts and/or co-localizes with costameric proteins such as dystrophin and dystrophin-associated proteins, integrin/vinculin/talin complex, α-actinin, desmin, and caveolin 3 (Oh et al., 2003). (As mentioned in advance #1, costameres are membrane-associated supramolecular structures that mediate muscle fiber adhesion to the extracellular matrix.) During skeletal muscle development, supervillin is located at the ends of differentiating myotubes, where it is thought to contribute to the myotendinous junction formation (Oh et al., 2003). In addition, supervillin plays a role in the myofibril assembly (Lee et al., 2007) and helps anchor peripheral myofibrils to the sarcolemma by connecting a Z-disc protein nebulin to costameric proteins dystrophin and γ-sarcoglycan (Lee et al., 2008; Spinazzola et al., 2015). (Highlighting the critical importance of myofibril anchoring, Z-discs of peripheral myofibrils are also linked to the sarcolemma through binding of a Z-disc protein filamin C to costameric proteins γ-sarcoglycan and integrin; reviewed in Peter et al., 2011.) Mutations in genes that encode costameric proteins mostly lead to muscular dystrophies, while mutations in myofibrillar and cytoskeletal proteins generally result in congenital myopathies; loss-of-function mutations in supervillin, which have previously not been associated with any muscle disease, are now known to cause a unique structural myopathy (Hedberg-Oldfors et al., 2020). The authors described four patients from two unrelated consanguineous families (two affected siblings in each family) that were ultimately shown to carry homozygous truncating mutations in supervillin, resulting in a complete loss of supervillin protein. While there were subtle clinical differences between these two families, shared clinical findings included onset in childhood/adolescence, muscle pain and stiffness without significant muscle weakness, wide neck and hypertrophy of back muscles, progressive contractures, moderately elevated CK levels, and mild cardiac involvement (mainly left ventricular hypertrophy). Muscle biopsies from all four patients showed frequent lobulated type 1 fibers, myofibrillar disarray (including occasional Z-band streaming and nemaline rods), subsarcolemmal protein aggregates, and autophagic vacuoles (Fig. 3; see also Hedberg-Oldfors et al., 2020). Aside from frequent lobulated fibers, these histopathologic features show some overlap with myofibrillar myopathies, a group of muscle disorders characterized by Z-disc instability and defects in the chaperone-assisted selective autophagy (reviewed in Margeta, 2020a); this histopathologic overlap may reflect the role of supervillin in the Z-disc stabilization / sarcomere anchoring. Interestingly, the currently known supervillin mutations are expected to abolish expression of all five supervillin isoforms (not just the archvillin isoform expressed in striated muscle); while these four patients did not show clinical involvement of any non-muscle tissues, the disease spectrum is likely to widen as additional patients (and mutations) are identified in the future. Going forward, it will be particularly interesting to elucidate how the loss of supervillin leads to fiber lobulation and to establish whether similar mechanisms operate in other myopathies with frequent lobulated fibers.
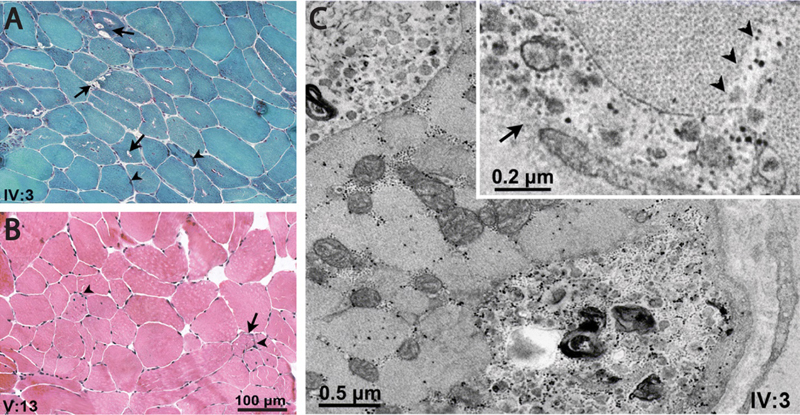
Figure 3. Histopathology of “supervillinopathy”. Biopsy from the deltoid muscle of Patient III:2 in Family 2, as visualized by light (A–C) and electron microscopy (D–G). A. A considerable number of partially atrophic lobulated muscle fibers with pointed, occasionally cap-like subsarcolemmal deposits (arrows) (H&E). B. Prominent oxidative enzyme activity in these fibers (arrows) (NADH). C. Atrophic lobulated fibers are almost exclusively type 1 fibers (arrows); no neurogenic pattern is observed (mATPase at pH 9.4). D. Subsarcolemmal accumulation of degraded myofibrils, glycogen, pleomorphic material and autophagy-associated (arrows) organelles. E. Heterogeneous lipoprotein deposits including maturing lipofuscin (arrows). F. Sporadically, nemaline rods are seen (arrow). G. Three rods in transverse section (arrows). (This figure and its legend were adopted from Figure 5 in Hedberg-Oldfors et al., 2020; this use is permitted under the Creative Commons Attribution 4.0 International License.) Advances in understanding of etiology and pathogenesis of neuromuscular diseases 5. Genetic vacuolar myopathies: Expanding etiology spectrum Vacuolar myopathies are pathologically defined by the presence of autophagic vacuoles that can be seen on the light microscopic level (“rimmed vacuoles”), electron microscopic level, or both. Vacuolar myopathies have varied etiologies that include myotoxic drugs, chronic inflammation, and genetic mutations; genetic forms can occur either as stand-alone myopathies or together with amyotrophic lateral sclerosis, frontotemporal lobar degeneration, and/or Paget’s disease of the bone as a component of multiple system proteinopathies (reviewed in Margeta, 2020a). Unsurprisingly, then, the differential diagnosis of a vacuolar myopathy remains broad, and the final diagnosis can be difficult to establish even in the era of widely available genetic testing; this complexity was highlighted by a 2020 study (Mair et al., 2020) that performed thorough clinicopathologic and genetic evaluation of 32 adult vacuolar myopathy cases from 30 unrelated families, and which includes a very comprehensive table of non-inflammatory causes that will be a useful resource for every practicing muscle pathologist. However, and highlighting the rapid pace of advancement in the NMD field, this table is already incomplete: a very exciting work published last year (Ruggieri et al., 2020) demonstrated that a repeat expansion in the lipid droplet-coating protein perilipin 4 is the cause of another rimmed vacuolar myopathy - the autosomal dominant distal myopathy that was previously linked to chromosome 19p13.3 (Di Blasi et al., 2004). The age at diagnosis in the originally published kindred of 19 individuals varied from 26 to 73 years, but was earlier in the later generations; the symptoms were variable, but never included dysphagia or dysphonia. Pathologically, the biopsies showed some unique features: vacuoles were mainly subsarcolemmal, membrane-limited, and sometimes connected to the cell surface and/or bordered by the basal lamina, but did not include filamentous inclusions and were not Congo red- or thioflavin S-positive (Fig. 4; see also Di Blasi et al., 2004; Ruggieri et al., 2020). In spite of early genetic linkage of this disease to chromosome 19p, the causative mutation proved challenging to elucidate; ultimately, Ruggieri et al. honed in on the correct target by performing quantitative mass spectroscopy of microdissected vacuoles, which identified perilipin 4 as the most enriched among 700 identified proteins. The subsequent analysis showed that the gene encoding perilipin 4, PLIN4, maps to 19p13.3; in affected patients, this gene contains a 891-nucelotide expansion of the repetitive nucleotide sequence in exon 4, resulting in extra 297 amino acids in the amphipathic domain of perilipin 4 protein.
Figure 4. Histopathology of “perilipinopathy” (19p13.3-linked distal myopathy). A-B. Gomori trichrome and H&E staining, respectively, in patients IV:3 and V:3, showing vacuoles (arrows) [rimmed, empty, or containing granular or basophilic material (arrowheads), mainly located in the subsarcolemmal region of fibers], fiber size variability, central nuclei, and mildly increased endomysial spaces. Note minimal changes in V:13. C. Electron micrographs unveiling vacuoles located in the subsarcolemmal region or deep in the sarcoplasm, containing small vesicles, membranous bodies, and granular debris; inset shows granular debris within a small subsarcolemmal vacuole (arrow) opening to the fiber’s surface and sarcolemmal interruption (arrowheads). (This figure and its legend were adopted from Figure 1 in Ruggieri et al., 2020; this use is permitted under the Creative Commons Attribution 4.0 International License.) Perilipins are proteins that localize to the surface of lipid droplets, where they interact with lipid enzymes and other regulators of lipid droplet metabolism; the five known isoforms all contain an 11-mer repeat sequence that repeats 3 times to form a 33-mer amphipathic helix required for lipid droplet localization (Copic et al., 2018). Perilipin 4 [the isoform most highly expressed in skeletal muscle, where it mainly localizes to the subsarcolemmal regions of type 1 fibers (Pourteymour et al., 2015)] has a very long amphipathic domain that normally consists of 29-31 33-mer repeats but is extended by 9 additional 33-mers in patients with 19p13.3 distal myopathy (Ruggieri et al., 2020). It is currently not understood how this expansion of the perilipin 4 amphipathic domain leads to muscle disease; however, the defect almost certainly does not involve metabolism of lipid droplets, which were similar in size and number in control and patient samples (Ruggieri et al., 2020). On the other hand, the patient samples showed an increase in subsarcolemmal perilipin 4 expression and co-localization of this protein with p62/SQSTM1, NBR1 and WDFY3 [autophagic adapters that play a key role in aggrephagy and granulophagy (selective autophagy of protein aggregates and stress granules, respectively)]. This finding provides a potential link between 19p13.3 distal myopathy and multiple system proteinopathies, which all show a defect in granulophagy (reviewed in Margeta, 2020a). Interestingly, expression of perilipin 4 in the CNS is regulated by a stress granule RNA-binding protein TIA1 (Heck et al., 2014), the gain-of-function mutations in which lead to Welander distal myopathy (Hackman et al., 2013; Klar et al., 2013). It remains to be shown whether TIA1 also regulates perilipin 4 expression in skeletal muscle and whether this regulation plays a role in pathogenesis of these two (and potentially other) distal vacuolar myopathies. 6. Vasculitic neuropathy secondary to eosinophilic granulomatosis with polyangiitis: Two pathogenetic mechanisms Vasculitic neuropathy, the most common indication for nerve biopsy in the current neuromuscular pathology practice, can be secondary to many different forms of vasculitis. Based on the most recent consensus nomenclature (Jennette et al., 2013), small vessel vasculitis (SVV; the category of vasculitis that is most likely to involve peripheral nerves) is divided into two subcategories – immune complex SVV (which shows abundant immunoglobulin deposition in the walls of affected vessels) and ANCA (antineutrophil cytoplasmic antibody)-associated SVV (where affected vessel walls show a paucity of immunoglobulin deposits). Systemic ANCA-associated SVV includes three distinct subcategories: microscopic polyangiitis, granulomatosis with polyangiitis (previously known as Wegener’s granulomatosis), and eosinophilic granulomatosis with polyangiitis (EGPA; previously known as Churg-Strauss syndrome). Interestingly, some patients with ANCA-associated SVV do not actually have detectable ANCA; it is not entirely clear whether that is because they have ANCA that cannot be detected with current methods, because they have novel, yet-to-be discovered ANCA, or because pathogenesis of their vasculitis actually does not involve ANCA at all (Jennette et al., 2013). A large clinicopathologic study of EGPA-associated neuropathy published last year (Nishi et al., 2020) sheds some light on this question by providing evidence for different pathogenic mechanisms in ANCA-positive and ANCA-negative forms of EGPA-associated neuropathy. EGPA is a form of systemic SVV that is associated with asthma, eosinophilia, and peripheral neuropathy; neuropathy is common and affects 50-75% of patients. Among the three subtypes of systemic ANCA-associated SVV, EGPA is least likely to show ANCA positivity: just 30-40% of patients have anti-myeloperoxidase ANCA, and almost none have anti-proteinase 3 ANCA (Nishi et al., 2020). To explore differences between ANCA-positive and ANCA-negative forms of EGPA, Nishi and co-authors have retrospectively investigated 82 consecutive patients who were diagnosed with EGPA-associated neuropathy and underwent sural nerve biopsy; 33% of study subjects were positive for anti-myeloperoxidase ANCA and none were positive for anti-proteinase 3 ANCA. Clinically, ANCA-positive and ANCA-negative EGPA patients were quite similar, although ANCA-positive patients were more likely to show involvement of the upper extremities than the ANCA-negative ones. Pathologically, both ANCA-positive and ANCA-negative cases showed predominantly axonal form of nerve damage with a focal or multifocal pattern of axon loss suggestive of ischemic injury; however, the vascular involvement differed between the two groups. Nerve biopsies from ANCA-positive patients were more likely to show frank vasculitis and/or destruction of vascular structures, with the mean diameter of affected vessels ~170 µm (Fig. 5A); in contrast, nerve biopsies from ANCA-negative cases were more likely to show a large number of eosinophils in the epineurial vessel lumina, epineurial vessels occluded by intraluminal eosinophils (with vessels smaller than 13 µm typically showing more than 50% occlusion; Fig. 5B), and extravasation of eosinophils into the endoneurium. Interestingly, the pattern of axon loss in ANCA-negative cases showed no topographic correlation with the location of endonurial eosinophils; rather, it was patchy (focal or multifocal), suggestive of nerve ischemia. Taken together, these findings suggest that there are two distinct pathogenetic mechanisms for ischemic injury in EGPA-associated neuropathy: frank vessel destruction in ANCA-positive cases and vessel occlusion by eosinophils in ANCA-negative cases (the latter mechanism is conceptually similar to vessel occlusion by intravascular large B-cell lymphoma, which causes tissue ischemia without directly destroying vascular structures). While both patterns of vessel involvement can be seen in skeletal muscle (Fig. 5) in addition to peripheral nerves of patients with EGPA, it remains to be seen whether differences between ANCA-positive and ANCA-negative EGPA cases will be conserved across all affected organ systems. In addition, it needs to be established whether similar mechanistic differences distinguish ANCA-positive and ANCA-negative cases in the other two types of ANCA-associated SVV. Finally, given the almost complete lack of vessel wall inflammation in ANCA-negative EGPA cases, it needs to be re-evaluated whether this clinicopathologic syndrome should remain classified as a vasculitis or whether a different disease category would be more appropriate.
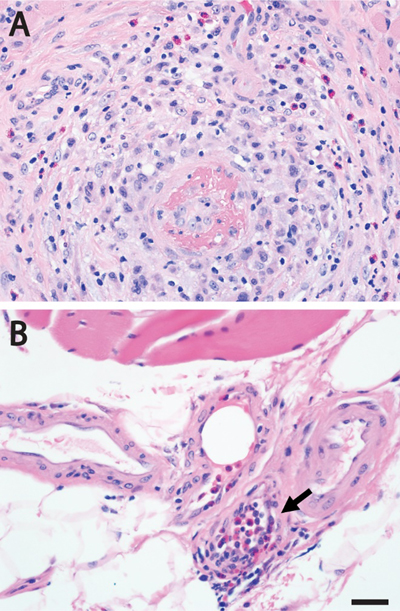
Figure 5. Two patterns of vascular involvement in eosinophilic granulomatosis with polyangiitis. A. Muscle biopsy from a patient with ANCA-positive EGPA shows necrotizing vasculitis affecting small intramuscular artery; the inflammatory infiltrate includes eosinophils. B. The lumen of a very small perimysial artery (arrow) is nearly completely occluded by intraluminal eosinophils in a muscle biopsy from a patient with EGPA; ANCA status was not reported. Representative images from H&E-stained sections of formalin-fixed, paraffin-embedded tissue are shown for both cases. Scale bar, 20 µm. 7. Spinal muscular atrophies: Skeletal muscle involvement Spinal muscular atrophies (SMAs) are a group of genetic disorders in which the loss of lower motor neurons (LMNs) in the anterior horn of the spinal cord, which typically occurs early in life, results in neurogenic muscle atrophy and weakness. The most common SMA subtype, 5q-SMA, accounts for ~95% of SMA cases and is caused by autosomal recessive, loss-of-function mutations in both copies of SMN1 (survival motor neuron 1) gene; the clinical phenotype is modified by the presence of the paralog gene, SMN2, which yields only a small number of full-length transcripts and is present in a variable number of copies in different individuals, with more copies resulting in a milder clinical phenotype and better survival. A different SMA subtype, known as SMALED (“spinal muscular atrophy lower extremity predominant”), is caused by autosomal dominant mutations in DYNC1H1 and BICD2 genes, which impair centripetal microtubular transport that moves intracellular cargo from the cell periphery to the perinuclear region [DYNC1H1 encodes a subunit of the dynamin/dynactin retrograde transport complex, while BICD2 encodes a cargo adapter protein that directly binds to DYNC1H1 (Koboldt et al., 2020)]. Pathogenesis of both 5q-SMA and SMALED was thought to involve loss of LMNs that occurs in a cell-autonomous manner; however, two studies published last year (Kim et al., 2020; Rossor et al., 2020) highlight the important and somewhat unexpected role of skeletal muscle in these disease processes. In addition to the LMN loss that is a defining feature of the SMA phenotype, BICD2 deficiency causes cerebellar hypoplasia through a pathway that involves Bergmann glia in a non-cell autonomous manner. To investigate whether the LMN loss in SMALED also involves non-cell autonomous mechanisms, Rossor et al. selectively deleted BICD2 gene from either LMNs or skeletal muscle (Rossor et al., 2020). While neither mouse model showed altered gait or decreased life expectancy, a subtle loss of small diameter (gamma) motor axons was observed in the mice lacking BICD2 in skeletal muscle; in contrast, there was no evident axon loss (or any other MN phenotype) in the mice lacking BICD2 expression in LMNs. Further experiments demonstrated that skeletal muscle from Bicd2-/- mice showed a reduction in the number of muscle spindles but no neurogenic changes indicative of the loss of alpha LMNs; similar findings were seen in mice lacking DYNC1H1. Finally, the authors used an in vitro assay to show a decreased secretory activity in fibroblasts from a SMALED patient with a BICD2 mutation when compared to fibroblasts from a control patient. Based on these findings, Rossor et al. proposed that the LMN loss in SMALED is non-cell autonomous and involves reduced neurotrophin secretion from skeletal muscle, which limits neurotrophin availability and leads to excess LMN apoptosis during development. While this hypothesis is intriguing, it is currently not clear whether a similar pathogenetic mechanism operates in human disease; in particular, biopsies from SMALED patients show a mixture of neurogenic and myopathic changes that is not well replicated by the existing mouse models. In addition, it is not clear why diminished neurotrophin availability would preferentially affect LMNs that innervate lower extremities. Nonetheless, the results of this study are sufficiently compelling to warrant further investigation. Kim et al. have used a similar experimental approach to explore the role of skeletal muscle in the 5q-SMA pathogenesis (Kim et al., 2020). Intriguingly, they found that the lack of SMN expression in skeletal muscle results in an age-dependent myopathy that is usually obscured by the concurrent neurogenic process driven by the LMN loss. In the presence of a single copy of SMN2 gene, SMN1 deficiency in skeletal muscle led to an early-onset myopathy and severely shortened life span, with most mice dying by the postnatal day 25. A milder phenotype was seen in mice deficient for SMN1 that carried two copies of SNM2 gene: at a young age, their skeletal muscles appeared normal but showed impaired regeneration following a myotoxic insult; by 6-7 months of age, outright myopathic changes and impaired functional muscle performance were observed, leading to early mortality with only 14% of mutants (but 95% of controls) viable at 18 months. Importantly, restoration of muscle SMN levels in 7-month-old mice mitigated the observed muscle pathology. Conceptually, this study reinforces the paradigm observed with many genetic diseases: a treatment that is effective but targets only the most severely affected organ system typically unmasks the effects of the same mutation on other organ systems that were previously not apparent, but now need to be addressed. For patients with 5q-SMA, the implications are more practical: nusinersen treatment, which is given intrathecally and therefore increases SMN expression in the spinal cord but not in skeletal muscle, may need to be combined with (or replaced by) a treatment that also increases SMN levels in peripheral tissues; one candidate for such therapy is AAV9-SMN, an AAV9 (adeno-associated virus 9) vector carrying human SMN transgene that was approved for 5q-SMA treatment in 2019, but is currently restricted to patients younger than 2 years of age [for more information on emerging treatments for 5q-SMA, see the prior update in this article series (Margeta, 2020b)]. Additional clinical trials that specifically focus on myopathic pathology in 5q-SMA will be needed to address this clinically important issue. Moreover, it will be important to determine how SMN deficiency leads to the failure of muscle maintenance, which seems to underpin this late-onset muscle pathology. 8. Idiopathic inflammatory myopathies: The role of autoantibodies targeting sarcolemmal repair proteins Based on the current clinicoseropathologic criteria, idiopathic inflammatory myopathies (IIMs) are divided into four distinct subcategories [dermatomyositis (DM), anti-synthetase syndrome-associated myositis (ASM), immune-mediated necrotizing myopathy (IMNM), and sporadic inclusion body myositis (sIBM); for a brief overview of the diagnostic criteria and other recent advances in the IIM field, see last year’s update in this article series (Margeta, 2020b)]. IIM pathogenesis is complex and only partly understood, but an interesting study published last year uncovered a feed-forward mechanism that contributes to IIM progression by linking development of autoantibodies against TRIM72 (also known as mitsugumin 53) with defective sarcolemmal repair and ongoing exposure of intracellular autoantigens (McElhanon et al., 2020). TRIM72 is a 53 kDa member of the TRIM family of proteins; it is highly expressed in striated muscle and has many different cellular functions, including a key role in the plasma membrane repair. Following sarcolemmal injury, TRIM72 nucleates and then coats intracellular lipid vesicles that accumulate at the site of the membrane break; fusion of these vesicles with damaged sarcolemma creates a “patch” that seals the membrane defect in a process that requires Ca2+ as well as interaction of TRIM72 with dysferlin and caveolin 3 (reviewed in Benissan-Messan et al., 2020). While mutations in dysferlin and caveolin 3 lead to limb-girdle muscular dystrophies 2B and 1C, respectively, mutations in TRIM72 have not yet been linked to any human disease; however, the myocardium of TRIM72-deficient mice is more vulnerable to ischemia (Cao et al., 2010), highlighting the importance of this protein for striated muscle integrity. Building on that body of work, McElhanon et al. have now implicated TRIM72 in the IIM pathogenesis by using an adaptive transfer animal model of IIM. In this model, lymph node cells from Syt7-/- FoxP3-/γ double mutant mice (which have defective membrane repair and are prone to autoimmunity because they lack Tregs) are transferred into immunologically naïve Rag1-/- mice, who one week after transfer develop an inflammatory myopathy. Intriguingly, endomysial inflammatory infiltrates composed of T cells and macrophages were present only in the proximal muscles of the recipient mice, while “leaky” muscle fibers with compromised sarcolemma were identified in both proximal and distal muscles. The presence of myofiber damage in non-inflamed distal muscles raised the possibility that humoral rather than cellular autoimmunity caused impaired sarcolemmal repair in this mouse model, leading the authors to investigate whether the recipient mice developed autoantibodies against one or more proteins involved in this process. Indeed, anti-TRIM72 autoantibodies were detectable in the sera of the recipient mice as well as in 10-30% (depending on the chosen cutoff) of sera from 103 human IIM patients. Importantly, the authors showed that anti-TRIM72 antibodies were pathogenic: treatment of isolated normal muscle fibers by either purified anti-TRIM72 antibodies or by human IIM sera positive for these antibodies caused a membrane repair defect (as measured by an in vitro membrane resealing assay), while depletion of anti-TRIM72 antibodies from human IIM sera strongly attenuated this effect. Based on these findings, the authors proposed a new model of IIM pathogenesis, in which sarcolemmal disruption caused by any number of events (including a viral or bacterial infection, trauma, or overexertion) results in exposure of sarcolemmal repair proteins to the immune system; in susceptible individuals, this exposure leads to development of autoantibodies that target TRIM72 and/or other repair proteins, compromising membrane resealing and leading to continuous autoantigen exposure in a feed-forward loop. While this model needs to be experimentally tested, it does provide possible explanation for association of COVID-19 (and other viral illnesses) with immune-mediated muscle pathology (discussed in advance #3). One limitation of this otherwise very nicely done study is the outdated classification of the patient IIM sera used for experiments (they were classified into DM and polymyositis categories, presumably because they were obtained from a specimen bank that performed specimen collection before introduction of the current IIM diagnostic criteria). Nevertheless, the study included information about the status of a subset of myositis-specific antibodies in the same IIM specimen set, providing at least partial clues to more up-to-date classification. Based on this limited information, anti-TRIM72 antibodies were present in a significant fraction of IIM patients with DM-associated antibodies (anti-Mi-2, anti-TIF1γ, and anti-SEA), as well as one of two patients with IMNM-associated anti-SRP antibodies; in contrast, they were not detected in any of patients with the ASM-associated anti-Jo-1 antibodies. While these findings need to be replicated and extended by evaluation of patients from all currently defined IIM subcategories (including sIBM and other subtypes of IMNM), they do raise the possibility that this new pathomechanism plays a bigger role in pathogenesis of DM and IMNM than ASM. In that context, it is intriguing that TIF1γ / TRIM33 – which is targeted by one of the DM-associated myositis-specific antibodies – is another member of the TRIM protein family, although in contrast to TRIM72 it does not seem to play a significant role in skeletal muscle repair (Parks et al., 2019). Advances in neuromuscular disease diagnostics 9. Large-scale electron microscopy Transmission electron microscopy (TEM) remains an important diagnostic tool in the contemporary neuromuscular pathology workflow, but some practical limitations diminish its potential usefulness. In particular, it is typically necessary to maximize the use of these expensive instruments while minimizing the number of people who must obtain specialized training required to operate them; as a result, diagnostic ultrastructural evaluation is typically performed on static images of select areas of interest that were previously captured by the trained laboratory staff. While reasonably effective, this approach makes it challenging to relate the captured nanoscale images to the microscale tissue architecture appreciated by light microscopy; in addition, depending on the staff expertise level, there is always a possibility that the captured images are not fully representative of the underlying pathology, leading to diagnostic errors. To circumvent some of these limitations, large-scale EM [also known as “virtual EM” or nanotomy (short for nanometer-scale anatomy)] has been developed over the last decade; while the first use of this technique in the NMD field was reported in 2003 (Sullivan et al., 2003), a wider adoption did not happen until last year (Dittmayer et al., 2020b; Dittmayer et al., 2020c; Rocha et al., 2020). In large-scale EM, the entire ultrathin sections are digitized by automated acquisition of multiple overlapping image tiles that are then stitched into a single large image; the resulting high-resolution digital images can then be evaluated on a standard computer workstation, enabling easy navigation between micrometer and nanometer scales across the entire ultrathin section. Initial applications of this technology were based on stitching of multiple traditionally acquired TEM images (Sullivan et al., 2003; Faas et al., 2012; Lee and Mak, 2011); while providing a proof-of-principle for virtual EM as a diagnostic and research tool, this approach had two major limitations - (1) imperfect image quality due to interference from the specimen support grids and various preparation flaws, and (2) a long time required to capture and stich a very large number of standard TEM images. In the years that followed, these limitations were addressed by replacing traditional TEM by field-emission scanning EM (FESEM); one FESEM image captures the field of view that is equivalent to ~100 TEM images, thus significantly reducing the amount of required image stitching and accelerating the entire process (Kuipers et al., 2016). In FESEM, imaging of ultrathin sections can be performed using a conductive silicon substrate that does not cause any visual interference, producing very sharp TEM-like images through detection of backscattered electrons (Dittmayer et al., 2018); this solves problem 1, but not entirely problem 2 because imaging speed remains slow (Kuipers et al., 2016). Alternatively, FESEM can be used for digitization of sections prepared with conventional TEM slot grids and imaged in a transmission mode with a scanning transmission electron microscopy (STEM) detector (Kuipers et al., 2016); this solves problem 2 due to faster imaging, but preparation of samples with minimal flaws remains challenging. Despite remaining limitations, the capabilities of this new technology are already impressive: if STEM is combined with conventional modern systems, automated digitization of entire ultrathin sections at a 7 nm-pixel resolution can be performed in about 12 h (Dittmayer et al., 2020c), while specialized high-throughput imaging systems based on FESEM and TEM technology can reduce the imaging time to about 10-20 min. When will large-scale EM become available for routine diagnostic use? That is difficult to predict, given that the most sophisticated version of the technique requires purchase of new, more advanced EM instruments; however, the cost could be justified by anticipated improvements in diagnostic accuracy, opportunities for digital pathology research, and reduction in the time spent on image acquisition by the laboratory staff. In the meantime, one can get a taste of this exciting new technology by viewing open-source datasets available at http://www.nanotomy.org/ Advances in neuromuscular disease treatment 10. Long-term efficacy and safety: Gene-silencing therapies for transthyretin amyloid neuropathy Transthyretin amyloidosis (ATTR) is caused by deposition of either wild-type or mutant transthyretin (TTR) in different tissues throughout the body; these two disease forms are referred to as ATTRwt and ATTRv (“v” for variant), respectively. TTR functions as a carrier for thyroxin and retinol/vitamin A and is primarily synthetized by the liver; clinical symptoms of ATTR are largely due to accumulation of TTR amyloid in the heart and peripheral nerves, but skeletal muscle can also be affected in rare cases (Fig. 6 and Pinto et al., 2020; see also Fig. 1 in Lam et al., 2015). Interestingly, recent work has shown that tissue damage in ATTR is caused not only by the amyloid fibril deposition but also by toxicity of nonfibrillar transthyretin oligomers, which directly damage endothelial cells and nerve fibers (reviewed in Koike and Katsuno, 2020).
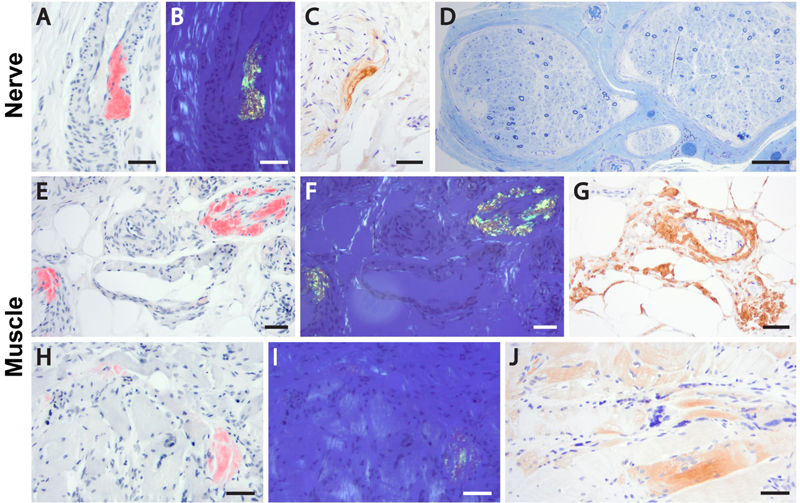
Figure 6. Transthyretin amyloid deposition in a peripheral nerve and skeletal muscle of a patient with ATTRv. Sural nerve biopsy (A-D) shows accumulation of Congo red-positive amyloid substance in the arterial walls (A); the deposits demonstrate apple-green birefringence under the polarized light (B) and are immunoreactive with anti-transthyretin antibody (C). Marked loss of myelinated axons is evident on Toluidine blue stain (D). Sections from a gastrocnemius muscle biopsy (E-J) show deposition of TTR amyloid in the arterial walls (E-G) and fiber sarcoplasm (H-J); well-developed neurogenic changes were also present (not shown). Subsequent to the biopsy diagnosis of TTR amyloid neuropathy and myopathy, the patient was found to carry a pathogenic TTR p.Phe84Leu mutation and was diagnosed with ATTRv; he was treated by inotersen and initially stabilized, but then continued to progress and will likely be switched to the patisiran therapy going forward. Panels A-C and E-J, formalin-fixed, paraffin embedded tissue; panel D, Epon-embedded tissue. Panels A-B, E-F, and H-I, Congo red stain; panels C, G and J, transthyretin immunoperoxidase stain; panel D, Toluidine blue stain. Scale bars, 50 µm. Fully assembled TTR is a conformationally stable homotetramer; in contrast, individual TTR monomers are prone to misfolding and aggregation. Thus, the currently approved ATTRv therapies aim either to stabilize the tetrameric quaternary structure of TTR (tafimidis or diflunisal; approved in the early 2010s) or to inhibit TTR synthesis through gene silencing approaches (inotersen or patisiran; both approved in 2018). Untreated ATTRv is a rapidly progressive disease with a poor prognosis, and the availability of several disease-modifying treatments was an incredible breakthrough for the NMD field. At the same time, many important treatment-related questions remained open, and had to be addressed through post-approval studies. For example, at the time of approval little was known about the long-term efficacy and safety of TTR-silencing drugs. In addition, it was not clear how different ATTRv treatment options compare to each other. While much work still remains to be done, at least some of these questions have been decisively answered by studies published in 2020. Inotersen is an antisense oligonucleotide that inhibits TTR production and thereby slows the progression of ATTRv polyneuropathy; based on a 2-year update from the open-label extension of the NEURO-TTR clinical trial (which led to its initial approval), no new safety concerns were identified and the drug continued to demonstrate therapeutic benefits. However, the outcomes were significantly better in study subjects who were originally placed in the experimental arm than in those who were originally in the placebo arm, indicating that early treatment is critical for inotersen efficacy (Brannagan et al., 2020). Similar results were seen with patisiran, which is an RNA-interference therapeutic that also inhibits TTR synthesis; a 12-month update from the open-label extension of the APOLLO clinical trial (which led to initial approval of this drug) also demonstrated no new safety concerns and continued efficacy, with sustained benefit in patients who were originally part of the patisiran arm and a new improvement in patients who were originally part of the placebo arm. Interestingly, the frequency of deaths was higher in the initial placebo group than in the initial patisiran group, again highlighting the benefits of early treatment (Adams et al., 2021). Finally, an indirect comparison of the data from NEURO-TTR and APOLLO clinical trials demonstrated that patisiran was generally more effective than inotersen, with a greater treatment benefit observed on all polyneuropathy and quality of life measures evaluated by both trials (Gorevic et al., 2021). Taken together with a prior study that showed a greater efficacy of inotersen compared to tafimidis in patients with ATTRv polyneuropathy (Plante-Bordeneuve et al., 2019), these data suggest that patisiran is the best currently available treatment for ATTRv polyneuropathy. It needs to be noted that both these indirect comparison studies were funded by Alnylam Pharmaceuticals, the manufacturer of patisiran; however, given the magnitude and robustness of the observed therapeutic differences, it is unlikely that the results were significantly confounded by the sponsor’s financial interests. Despite these advances, a lot remains to be explored. For example, it needs to be established whether TTR-silencing therapies will also be effective for treatment of ATTRwt polyneuropathy, and - if they are - whether their therapeutic benefit will be superior to that of tafimidis (which has been approved for ATTRwt treatment). Similarly, both TTR-silencing drugs still need to be evaluated as treatments for ATTRv (and ATTRwt) cardiomyopathy: initial exploratory studies showed that inotersen led to no improvement in cardiac function while patisiran stopped (and even reversed) the progression of cardiac disease in ATTRv patients (reviewed in Adams and Slama, 2020), but randomized clinical trials addressing the cardiac aspect of ATTR still need to be done. Disclosure statement The author receives research support from Audentes Therapeutics as a member of the muscle biopsy review committee for the ASPIRO clinical trial (NCT03199469), which is evaluating the safety and efficacy of gene transfer therapy for X-linked myotubular myopathy. Acknowledgements I am grateful to Ms. Christine Lin for assistance with figure preparation. References Adams, D., Polydefkis, M., Gonzalez-Duarte, A., Wixner, J., Kristen, A.V., Schmidt, H.H., Berk, J.L., Losada Lopez, I.A., Dispenzieri, A., Quan, D., et al. (2021). Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol 20, 49-59. Adams, D., and Slama, M. (2020). Hereditary transthyretin amyloidosis: current treatment. Curr Opin Neurol 33, 553-561. Anklesaria, Z., Frankman, J., Gordin, J., Zhan, J., and Liu, A.K. (2020). Fatal rhabdomyolysis in a COVID-19 patient on rosuvastatin. Cureus 12, e11186. Benissan-Messan, D.Z., Zhu, H., Zhong, W., Tan, T., Ma, J., and Lee, P.H.U. (2020). Multi-cellular functions of MG53 in muscle calcium signaling and regeneration. Front Physiol 11, 583393. Brannagan, T.H., Wang, A.K., Coelho, T., Waddington Cruz, M., Polydefkis, M.J., Dyck, P.J., Plante-Bordeneuve, V., Berk, J.L., Barroso, F., Merlini, G., et al. (2020). Early data on long-term efficacy and safety of inotersen in patients with hereditary transthyretin amyloidosis: a 2-year update from the open-label extension of the NEURO-TTR trial. Eur J Neurol 27, 1374-1381. Buckholz, A.P., Kaplan, A., Rosenblatt, R.E., and Wan, D. (2020). Clinical Characteristics, Diagnosis, and Outcomes of 6 Patients With COVID-19 Infection and Rhabdomyolysis. Mayo Clin Proc 95, 2557-2559. Cagnazzo, F., Arquizan, C., Derraz, I., Dargazanli, C., Lefevre, P.H., Riquelme, C., Gaillard, N., Mourand, I., Gascou, G., Bonafe, A., et al. (2020). Neurological manifestations of patients infected with the SARS-CoV-2: a systematic review of the literature. J Neurol, ePub ahead of print. Cao, C.M., Zhang, Y., Weisleder, N., Ferrante, C., Wang, X., Lv, F., Zhang, Y., Song, R., Hwang, M., Jin, L., et al. (2010). MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 121, 2565-2574. Caress, J.B., Castoro, R.J., Simmons, Z., Scelsa, S.N., Lewis, R.A., Ahlawat, A., and Narayanaswami, P. (2020). COVID-19-associated Guillain-Barre syndrome: The early pandemic experience. Muscle Nerve 62, 485-491. Copic, A., Antoine-Bally, S., Gimenez-Andres, M., La Torre Garay, C., Antonny, B., Manni, M.M., Pagnotta, S., Guihot, J., and Jackson, C.L. (2018). A giant amphipathic helix from a perilipin that is adapted for coating lipid droplets. Nat Commun 9, 1332. Dalakas, M.C. (2020). Guillain-Barre syndrome: The first documented COVID-19-triggered autoimmune neurologic disease: More to come with myositis in the offing. Neurol Neuroimmunol Neuroinflamm 7, e781. De Sanctis, P., Doneddu, P.E., Vigano, L., Selmi, C., and Nobile-Orazio, E. (2020). Guillain-Barre syndrome associated with SARS-CoV-2 infection. A systematic review. Eur J Neurol 27, 2361-2370. Di Blasi, C., Moghadaszadeh, B., Ciano, C., Negri, T., Giavazzi, A., Cornelio, F., Morandi, L., and Mora, M. (2004). Abnormal lysosomal and ubiquitin-proteasome pathways in 19p13.3 distal myopathy. Ann Neurol 56, 133-138. Ding, Y., Wang, H., Shen, H., Li, Z., Geng, J., Han, H., Cai, J., Li, X., Kang, W., Weng, D., et al. (2003). The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 200, 282-289. Dittmayer, C., Meinhardt, J., Radbruch, H., Radke, J., Heppner, B.I., Heppner, F.L., Stenzel, W., Holland, G., and Laue, M. (2020a). Why misinterpretation of electron micrographs in SARS-CoV-2-infected tissue goes viral. Lancet 396, e64-e65. Dittmayer, C., Siegert, E., Uruha, A., Goebel, H., and Stenzel, W. (2020b). Large-scale electron microscopy reveals capillary pathology in muscle samples of patients with systemic sclerosis. Neuromuscul Disord 30, S150. Dittmayer, C., Stenzel, W., Goebel, H.H., Krusche, M., Schneider, U., Uruha, A., and Englert, B. (2020c). Morphological characteristics of the transition from juvenile to adult dermatomyositis. Neuropathol Appl Neurobiol 46, 790-794. Dittmayer, C., Volcker, E., Wacker, I., Schroder, R.R., and Bachmann, S. (2018). Modern field emission scanning electron microscopy provides new perspectives for imaging kidney ultrastructure. Kidney Int 94, 625-631. Faas, F.G., Avramut, M.C., van den Berg, B.M., Mommaas, A.M., Koster, A.J., and Ravelli, R.B. (2012). Virtual nanoscopy: generation of ultra-large high resolution electron microscopy maps. J Cell Biol 198, 457-469. Ferrandi, P.J., Alway, S.E., and Mohamed, J.S. (2020). The interaction between SARS-CoV-2 and ACE2 may have consequences for skeletal muscle viral susceptibility and myopathies. J Appl Physiol (1985) 129, 864-867. Filosto, M., Cotti Piccinelli, S., Gazzina, S., Foresti, C., Frigeni, B., Servalli, M.C., Sessa, M., Cosentino, G., Marchioni, E., Ravaglia, S., et al. (2020). Guillain-Barre syndrome and COVID-19: an observational multicentre study from two Italian hotspot regions. J Neurol Neurosurg Psychiatry, ePub ahead of print. Fragiel, M., Miro, O., Llorens, P., Jimenez, S., Pinera, P., Burillo, G., Martin, A., Martin-Sanchez, F.J., Garcia-Lamberechts, E.J., Jacob, J., et al. (2020). Incidence, clinical, risk factors and outcomes of Guillain-Barre in Covid-19. Ann Neurol, ePub ahead of print. Gorevic, P., Franklin, J., Chen, J., Sajeev, G., Wang, J.C.H., and Lin, H. (2021). Indirect treatment comparison of the efficacy of patisiran and inotersen for hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert Opin Pharmacother 22, 121-129. Hackman, P., Sarparanta, J., Lehtinen, S., Vihola, A., Evila, A., Jonson, P.H., Luque, H., Kere, J., Screen, M., Chinnery, P.F., et al. (2013). Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol 73, 500-509. Heck, M.V., Azizov, M., Stehning, T., Walter, M., Kedersha, N., and Auburger, G. (2014). Dysregulated expression of lipid storage and membrane dynamics factors in Tia1 knockout mouse nervous tissue. Neurogenetics 15, 135-144. Hedberg-Oldfors, C., Meyer, R., Nolte, K., Abdul Rahim, Y., Lindberg, C., Karason, K., Thuestad, I.J., Visuttijai, K., Geijer, M., Begemann, M., et al. (2020). Loss of supervillin causes myopathy with myofibrillar disorganization and autophagic vacuoles. Brain 143, 2406-2420. Hooper, J.E., Uner, M., Priemer, D.S., Rosenberg, A., and Chen, L. (2020). Muscle biopsy findings in a case of SARS-CoV-2-associated muscle injury. J Neuropathol Exp Neurol, ePub ahead of print. Jennette, J.C., Falk, R.J., Bacon, P.A., Basu, N., Cid, M.C., Ferrario, F., Flores-Suarez, L.F., Gross, W.L., Guillevin, L., Hagen, E.C., et al. (2013). 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65, 1-11. Keddie, S., Pakpoor, J., Mousele, C., Pipis, M., Machado, P.M., Foster, M., Record, C.J., Keh, R.Y.S., Fehmi, J., Paterson, R.W., et al. (2020). Epidemiological and cohort study finds no association between COVID-19 and Guillain-Barre syndrome. Brain, ePub ahead of print. Kim, J.K., Jha, N.N., Feng, Z., Faleiro, M.R., Chiriboga, C.A., Wei-Lapierre, L., Dirksen, R.T., Ko, C.P., and Monani, U.R. (2020). Muscle-specific SMN reduction reveals motor neuron-independent disease in spinal muscular atrophy models. J Clin Invest 130, 1271-1287. Klar, J., Sobol, M., Melberg, A., Mabert, K., Ameur, A., Johansson, A.C., Feuk, L., Entesarian, M., Orlen, H., Casar-Borota, O., et al. (2013). Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum Mutat 34, 572-577. Koboldt, D.C., Waldrop, M.A., Wilson, R.K., and Flanigan, K.M. (2020). The genotypic and phenotypic spectrum of BICD2 variants in spinal muscular atrophy. Ann Neurol 87, 487-496. Koike, H., and Katsuno, M. (2020). Transthyretin amyloidosis: Update on the clinical spectrum, pathogenesis, and disease-modifying therapies. Neurol Ther 9, 317-333. Kuipers, J., Kalicharan, R.D., Wolters, A.H., van Ham, T.J., and Giepmans, B.N. (2016). Large-scale scanning transmission electron microscopy (nanotomy) of healthy and injured zebrafish brain. J Vis Exp 111, 53635. Lam, L., Margeta, M., and Layzer, R. (2015). Amyloid polyneuropathy caused by wild-type transthyretin. Muscle Nerve 52, 146-149. Lee, K.C., and Mak, L.S. (2011). Virtual electron microscopy: a simple implementation creating a new paradigm in ultrastructural examination. Int J Surg Pathol 19, 570-575. Lee, M.A., Joo, Y.M., Lee, Y.M., Kim, H.S., Kim, J.H., Choi, J.K., Ahn, S.J., Min, B.I., and Kim, C.R. (2008). Archvillin anchors in the Z-line of skeletal muscle via the nebulin C-terminus. Biochem Biophys Res Commun 374, 320-324. Lee, Y.M., Kim, H.S., Choi, J.H., Choi, J.K., Joo, Y.M., Ahn, S.J., Min, B.I., and Kim, C.R. (2007). Knockdown of Archvillin by siRNA Inhibits Myofibril Assembly in Cultured Skeletal Myoblast. J Exp Biomed Sci 13, 251-261. Lentscher, A.J., McCarthy, M.K., May, N.A., Davenport, B.J., Montgomery, S.A., Raghunathan, K., McAllister, N., Silva, L.A., Morrison, T.E., and Dermody, T.S. (2020). Chikungunya virus replication in skeletal muscle cells is required for disease development. J Clin Invest 130, 1466-1478. Leung, T.W., Wong, K.S., Hui, A.C., To, K.F., Lai, S.T., Ng, W.F., and Ng, H.K. (2005). Myopathic changes associated with severe acute respiratory syndrome: a postmortem case series. Arch Neurol 62, 1113-1117. Mair, D., Biskup, S., Kress, W., Abicht, A., Bruck, W., Zechel, S., Knop, K.C., Koenig, F.B., Tey, S., Nikolin, S., et al. (2020). Differential diagnosis of vacuolar myopathies in the NGS era. Brain Pathol 30, 877-896. Manzano, G.S., Woods, J.K., and Amato, A.A. (2020). Covid-19-associated myopathy caused by type I interferonopathy. N Engl J Med 383, 2389-2390. Mao, L., Jin, H., Wang, M., Hu, Y., Chen, S., He, Q., Chang, J., Hong, C., Zhou, Y., Wang, D., et al. (2020). Neurologic manifestations of hospitalized patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol 77, 683-690. Margeta, M. (2020a). Autophagy defects in skeletal myopathies. Annu Rev Pathol 15, 261-285. Margeta, M. (2020b). Top ten discoveries of the year: Neuromuscular disease. Free Neuropathol 1, 4. McElhanon, K.E., Young, N., Hampton, J., Paleo, B.J., Kwiatkowski, T.A., Beck, E.X., Capati, A., Jablonski, K., Gurney, T., Perez, M.A.L., et al. (2020). Autoantibodies targeting TRIM72 compromise membrane repair and contribute to inflammatory myopathy. J Clin Invest 130, 4440-4455. Nishi, R., Koike, H., Ohyama, K., Fukami, Y., Ikeda, S., Kawagashira, Y., Iijima, M., Katsuno, M., and Sobue, G. (2020). Differential clinicopathologic features of EGPA-associated neuropathy with and without ANCA. Neurology 94, e1726-e1737. Oh, S.W., Pope, R.K., Smith, K.P., Crowley, J.L., Nebl, T., Lawrence, J.B., and Luna, E.J. (2003). Archvillin, a muscle-specific isoform of supervillin, is an early expressed component of the costameric membrane skeleton. J Cell Sci 116, 2261-2275. Parks, C.A., Pak, K., Pinal-Fernandez, I., Huang, W., Derfoul, A., and Mammen, A.L. (2019). Trim33 (Tif1gamma) is not required for skeletal muscle development or regeneration but suppresses cholecystokinin expression. Sci Rep 9, 18507. Peter, A.K., Cheng, H., Ross, R.S., Knowlton, K.U., and Chen, J. (2011). The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog Pediatr Cardiol 31, 83-88. Pinto, M.V., Milone, M., Mauermann, M.L., Dyck, P.J.B., Alhammad, R., McPhail, E.D., Grogan, M., and Liewluck, T. (2020). Transthyretin amyloidosis: Putting myopathy on the map. Muscle Nerve 61, 95-100. Plante-Bordeneuve, V., Lin, H., Gollob, J., Agarwal, S., Betts, M., Fahrbach, K., Chitnis, M., and Polydefkis, M. (2019). An indirect treatment comparison of the efficacy of patisiran and tafamidis for the treatment of hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert Opin Pharmacother 20, 473-481. Pourteymour, S., Lee, S., Langleite, T.M., Eckardt, K., Hjorth, M., Bindesboll, C., Dalen, K.T., Birkeland, K.I., Drevon, C.A., Holen, T., et al. (2015). Perilipin 4 in human skeletal muscle: localization and effect of physical activity. Physiol Rep 3, e12481. Rocha, M.L., Dittmayer, C., Uruha, A., Korinth, D., Chaoui, R., Schlembach, D., Rossi, R., Pelin, K., Suk, E.K., Schmid, S., et al. (2020). A novel mutation in NEB causing foetal nemaline myopathy with arthrogryposis during early gestation. Neuromuscul Disord, ePub ahead of print. Rosato, C., Bolondi, G., Russo, E., Oliva, A., Scognamiglio, G., Mambelli, E., Longoni, M., Rossi, G., and Agnoletti, V. (2020). Clinical, electromyographical, histopathological characteristics of COVID-19 related rhabdomyolysis. SAGE Open Med Case Rep 8, 2050313X20983132. Rossor, A.M., Sleigh, J.N., Groves, M., Muntoni, F., Reilly, M.M., Hoogenraad, C.C., and Schiavo, G. (2020). Loss of BICD2 in muscle drives motor neuron loss in a developmental form of spinal muscular atrophy. Acta Neuropathol Commun 8, 34. Ruggieri, A., Naumenko, S., Smith, M.A., Iannibelli, E., Blasevich, F., Bragato, C., Gibertini, S., Barton, K., Vorgerd, M., Marcus, K., et al. (2020). Multiomic elucidation of a coding 99-mer repeat-expansion skeletal muscle disease. Acta Neuropathol 140, 231-235. Shang, M., Cappellesso, F., Amorim, R., Serneels, J., Virga, F., Eelen, G., Carobbio, S., Rincon, M.Y., Maechler, P., De Bock, K., et al. (2020). Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature 587, 626-631. Spinazzola, J.M., Smith, T.C., Liu, M., Luna, E.J., and Barton, E.R. (2015). Gamma-sarcoglycan is required for the response of archvillin to mechanical stimulation in skeletal muscle. Hum Mol Genet 24, 2470-2481. Sullivan, K.A., Brown, M.S., Harmon, L., and Greene, D.A. (2003). Digital electron microscopic examination of human sural nerve biopsies. J Peripher Nerv Syst 8, 260-270. Wang, K., Yaghi, O.K., Spallanzani, R.G., Chen, X., Zemmour, D., Lai, N., Chiu, I.M., Benoist, C., and Mathis, D. (2020). Neuronal, stromal, and T-regulatory cell crosstalk in murine skeletal muscle. Proc Natl Acad Sci U S A 117, 5402-5408. Willingham, T.B., Kim, Y., Lindberg, E., Bleck, C.K.E., and Glancy, B. (2020). The unified myofibrillar matrix for force generation in muscle. Nat Commun 11, 3722. Zhang, H., Charmchi, Z., Seidman, R.J., Anziska, Y., Velayudhan, V., and Perk, J. (2020). COVID-19-associated myositis with severe proximal and bulbar weakness. Muscle Nerve 62, E57-E60.
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |