|
|
|
Free Neuropathology 2:1 (2021) |
|
Review |
|
Neuroinflammation: 2021 update |
|
Hans Lassmann |
|
Center for Brain Research, Medical University of Vienna, Austria |
|
Corresponding author: |
|
Submitted: 18 December 2020 Accepted: 08 January 2021 Copyedited by: Aivi Nguyen Published: 12 January 2021 |
|
Keywords: Brain inflammation, COVID-19, Multiple sclerosis, Alzheimer’s disease, MOGAD |
|
Abstract Key requirements for the validity of a neuropathological study are the inclusion of large numbers of biopsy or autopsy cases and proper controls, the rigorous classification of the basic neuropathology and the selection of the most suitable technologies for investigation. Whether the studies are performed with the fanciest, new, and state of the art technology or with rather conventional methodology is of minor importance. Following these criteria, a spectrum of neuropathological studies has been published in 2020, which provides new insights on important questions related to neurological disease. They include the pathological substrate of brain disease in COVID-19 infected patients, the nature of the adaptive and innate inflammatory response, or the type and mechanisms of tissue injury and repair in multiple sclerosis, and diagnostically relevant or mechanistic new insights into antibody-mediated diseases of the central nervous system. Other studies describe in detail the dynamic changes of brain inflammation in patients with trisomy 21 as a disease model for Alzheimer’s disease, or the presence and consequences of vascular comorbidities in a chronic inflammatory disease, such as multiple sclerosis. All these contributions have provided important, highly relevant clues for basic and translational neuroscience. 1) What was the brain pathology in patients who died during the course of COVID-19? The COVID-19 pandemic has been a major challenge for society and particularly for health care institutions during the last year. Although COVID-19 primarily affects the respiratory system and the major cause of death is pneumonia with respiratory failure, other organs, such as the renal, cardiovascular system, or the digestive tract, are affected (Paterson et al. 2020). Most relevant for neuropathology is that the nervous system, too, can be a target, reflected for instance by anosmia and ageusia, encephalopathy, focal ischemic stroke, encephalitis, meningitis, or polyneuritis (Li et al. 2020, Paterson et al. 2020, Liu et al. 2020, Hernandez-Fernandez et al. 2020). To what extent these neurological deficits are due to direct SARS-Cov2 infection of the nervous tissue, immune-mediated brain damage in the course of a systemic cytokine storm, or are secondary complications of respiratory failure, the intensive care setting, or comorbidities is not clear (Liu et al. 2020). Neuropathology plays a critical role in answering these questions. The first approach to answer these questions was to analyze whether molecules, which are involved in cellular virus infection or propagation, are expressed within the nervous system. Angiotensin concerting enzyme 2 (ACE2) is one of the cellular receptors recognized by SARS-Cov2, when docking to the cell surface. Thus, the presence of ACE2 on brain cells, such as neurons, glia or cerebral endothelial cells suggests that brain infections is possible (Kabbani and Olds 2020, Kanwar et al. 2020). Another docking molecule for SARS-Cov2 is Neuropilin 1 (NRP1) and a high expression of this molecule has been found in virus infected cells in the nasal cavity in COVID-19 patients (Cantuti-Castelvetri et al. 2020). Early neuropathological studies in the COVID-19 pandemic were restricted to single case reports or investigations in very small patient cohorts, and they were generally restricted to some basic neuropathological investigations (Kantonen et al. 2020, Reichard et al. 2020, Jensen et al. 2020, Younger 2020, Liu et al. 2020). The results were diverse and controversial, mainly providing evidence for brain damage that was not directly linked to SARS-Cov2 infection. The first systematic study on this issue appeared on October 2020 in Lancet Neurology (Matschke et al. 2020), and this study combined classical neuropathology with virology and molecular studies on gene expression. In a first set of data the authors describe that different molecules, involved in virus docking and propagation, show a preferential expression in different cells of the central nervous system. For example, ACE2 was mainly found in oligodendrocytes. The highest expression of transmembrane protease serine subtype 2 and 4 (TMPRSS2 / 4) was seen in neurons, and NRP-1 was mainly present in endothelial cells and astrocytes. These data suggest different mechanisms of infection in different brain cells. The neuropathological studies documented the presence of focal and diffuse ischemic lesions and diffuse astrogliosis. Microglia activation with microglia nodules and inflammation were mainly seen in the lower brain stem. In about half of the patients, virus was found either by PCR or immunocytochemistry. However, the virus load was very low and by immunohistochemistry only a very small number of virus infected cells became apparent. This study is the first to describe the basic neuropathology seen in COVID-19 patients and is, thus, very important and groundbreaking. However, it also has important limitations. It is based on autopsies of 43 patients, which is likely not sufficient to cover the entire neuropathological spectrum of the disease. Another limitation is that it does not contain data regarding the neurological status of the patients. Thus, it remains unclear, to what extent patients with specific neurological disease manifestations have been included. The third limitation is that it does not include a proper patient control group and, particularly, patients with similarly severe systemic immune activation. This particular question has been addressed in another study on COVID-19 neuropathology, which revealed that the degree of inflammation and microglia activation seen in COVID-19 patients is similar to that of patients who died under septic conditions (Deigendesch et al. 2020), thus suggesting that it may at least in part be a secondary consequence of systemic immune activation. Another study, also based on a large sample of autopsies, mainly focused on the possible routes of CNS infection (Meinhardt et al. 2020). It shows high virus load in the olfactory epithelium, including the olfactory sensory neurons, and in the olfactory pathways within the CNS, supporting the view of a neuronal route of virus entry into the CNS. Additionally, however, virus antigen was also present in cerebral endothelial cells, associated with micro-thrombosis and cerebral microinfarcts. In summary, current pathological studies show that the central nervous system can be infected with SARS-Cov2, that local infection can be associated with nervous system damage, but the extent of infection is low. A large part of the neuropathological changes seems to be secondary to systemic immune activation and cytokine storm, critical-illness related encephalopathy, or hypoxia and comorbidities. 2) What new insights have emerged into phenotype and disease mechanisms of antibody mediated autoimmune diseases of the central nervous system? The discovery that autoantibodies against neuronal ion channels or neurotransmitter receptors are associated with a spectrum of acute and chronic neurological diseases, has revolutionized neurological disease research (Höftberger and Lassmann 2017). It has been shown that these diseases, which had before been regarded as functional diseases, neurodegenerative disorders or toxic conditions, are immune-mediated and can be successfully treated with immunosuppression. The number of diseases falling into this category has profoundly increased during the last years (Höftberger and Lassmann 2017). One reason, which may in part explain the increase in prevalence, is the introduction of immunological checkpoint inhibitor therapies in oncology. Immunological checkpoints are critical steps in T-cell activation, which prohibit the development of auto-reactive T-cells and auto-antibodies. When these checkpoints are inhibited, immune reactions against antigens of malignant neoplasms can be triggered, but this may occur at the expense of autoimmunity, which is frequently directed against neuronal antigens (Sechi et al. 2020). On this basis it is not surprising that auto-antibody-associated diseases of the central nervous system remained in the focus of research interest in 2020. The respective studies provided novel insights into the function of NMDA and glycine receptor directed antibodies in relation to the clinical disease spectrum (Matute et al. 2020, Carceles-Cordon et al. 2020, Rauschenberger et al. 2020) or how autoantibodies against IgLON 5 may trigger intracellular accumulation of tau-tangles (Landa et al. 2020). Here, I focus on one study, which deals with an interesting but also controversial aspect of these diseases. Pitsch et al. (2020) identified antibodies against the postsynaptic actin binding protein drebrin in patients with severe epileptic seizures, which were associated with encephalopathy and neuropsychiatric symptoms, including depression and cognitive impairment. The antibodies, identified by selective binding to the neuropil and by western blot and sequencing, were specific for these patients, and the specificity for the target antigen was proven by the lack of binding in drebrin knock out mice. Drebrin is an intracellular antigen, which is not exposed on the extracellular surface of neurons or synapses. For this reason, the antibodies themselves may not be pathogenic, but just represent a diagnostically useful marker for an autoimmune attack of cytotoxic T-cells, as it occurs in many classical paraneoplastic diseases. The pathology, described in a biopsy of one of the cases, is in line with this assumption, showing infiltrating T-cells in close contact with neurons. However, the study further describes neurophysiological experiments, which show that the antibodies induce altered neuronal activity patterns and increased firing and bursting rates in neuronal networks in vitro. These data suggest that auto-antibodies directed against a cytoplasmic protein in synapses may reach their specific target and induce functional changes. The authors suggest that extensive exocytosis and endocytosis, which takes place in active synapses, facilitates the entry of the antibodies into the intracellular compartment, but this proposed mechanism does not explain how the antibodies leave the endosomal compartment and access the cytosol. This important cell biological question with important disease relevance has to be clarified in future studies. 3) What is the pathological difference between MOG antibody associated inflammatory demyelinating disease (MOGAD) and multiple sclerosis (MS)? Focal plaques of demyelination with axonal preservation and reactive gliosis developing in the context of a chronic inflammatory reaction in the central nervous system has been regarded as the specific hallmark of MS pathology. However, a new disease entity has recently emerged, which is an inflammatory demyelinating disease associated with antibodies directed against myelin oligodendrocyte glycoprotein (Reindl and Waters 2019). Not all anti-MOG antibodies are pathogenic, but only those that are directed against a conformational epitope, which is accessible on the surface of native oligodendrocytes or on MOG-transfected cell lines. Using these diagnostic tests, it became clear that MOG antibody-associated disease (MOGAD) differs from MS by its clinical manifestation and course as well as by its response to immunomodulatory treatment (Fujihara and Cook 2020). To explain these clinical differences, a neuropathological comparison of these diseases is mandatory, but respective knowledge on MOGAD was restricted to few biopsies with very limited tissue samples. This changed in 2020, with two studies describing the neuropathological changes in large cohorts of MOGAD patients (Höftberger et al. 2020, Takai et al. 2020). The lesions in MOGAD differ from those seen in MS in many aspects, including their topographical distribution in the CNS, the type of demyelination, and the nature of the inflammatory response. In MS, new lesions are formed by simultaneous demyelination in large, focal areas or by the peripheral expansion of pre-existing lesions located throughout the brain and spinal cord. In contrast, MOGAD demyelination occurs by confluence of small perivenous lesions, generally resulting in a demyelination pattern similar to that seen in acute disseminated encephalomyelitis (Figure 1). Demyelination in MOGAD is associated with complement deposition at the site of active myelin injury, but the degree of complement activation is much less compared to that seen in patients with aquaporin 4 antibody associated neuromyelitis optica (NMO). In both conditions, MS as well as MOGAD, the basic lesion is characterized by inflammatory demyelination, partial axonal preservation and reactive astrocytic gliosis. However, the inflammatory reaction is fundamentally different. While in MS the dominant inflammatory reaction is seen around the larger drainage veins in the periventricular tissue and the meninges, in MOGAD the smaller veins and venules are mainly affected. Finally, in MOGAD, infiltrating lymphocytes are mainly CD4+ T-cells with low numbers of CD8+ T-cells and B-cells; the dominant lymphocytes in active MS lesions are tissue resident CD8+ effector memory T-cells and B-cells / plasma cells. All these data indicate that MOGAD and MS are fundamentally different diseases.
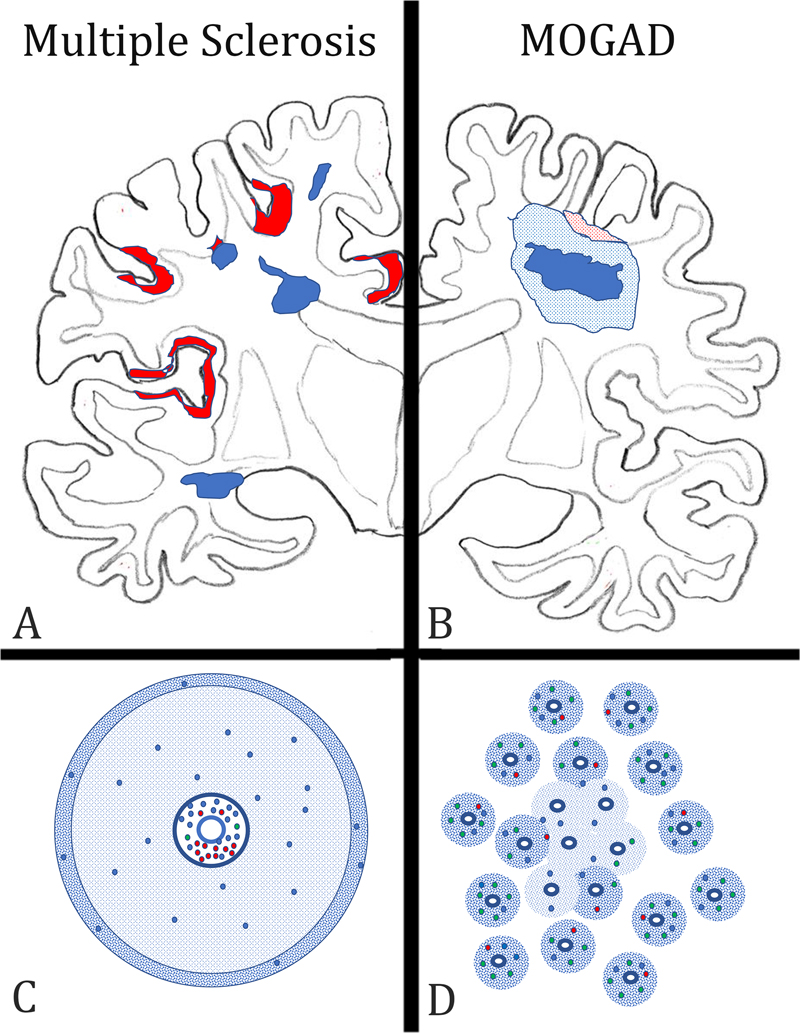
Figure 1: Key neuropathological features, distinguishing multiple sclerosis (MS) from the inflammatory demyelinating disease, which is associated with auto-antibodies against Myelin Oligodendrocyte Glycoprotein (MOGAD) in the forebrain. A) In MS, the lesions (blue areas) are located around large cerebral veins with a predilection site around the ventricle and the subcortical white matter. They are sharply demarcated from the surrounding peri-plaque white matter and frequently display finger like perivenous extensions into the deep white matter. In the cortex, band like subpial lesions (red areas) dominate, mainly located in the deep sulci of the cortical ribbon. B) In MOGAD, lesions are mainly located in the optic nerves and spinal cord, but their structural details have so far not been clearly outlined. When lesions are present in the hemispheres of the forebrain, a type of tissue injury is seen, which resembles acute disseminated encephalomyelitis. It consists of perivenous sleeves of demyelination around small veins and venules (blue dotted areas), which show confluence in the lesion center (dark blue area). When the cortex is affected small perivenous intra-cortical demyelination is seen most frequently (red dotted areas). C) In MS, the lesions form around larger veins with prominent perivascular spaces. The perivascular spaces contain mainly B-cells (red dots) and CD8+ T-lymphocytes (blue dots), the latter also disperse into the plaque parenchyma. CD4+ T-cells (green dots) are very sparse. A characteristic feature of active MS lesions is the radial expansion, reflected by a rim of activated macrophages at the lesion edge (blue dotted area). D) In contrast, in MOGAD, numerous small perivenous sheaths of demyelination are present, which arise around small veins and venules (blue dotted areas). In the inflammatory infiltrates the CD4+ T-cells dominate, while there is a much lower contribution of CD8+ T-cells (blue dots) or B-cells and plasma cells (red dots). Confluence of the perivenous lesions results in larger demyelinated plaques. Interestingly, the pathology of MOGAD closely resembles the pathology seen in experimental models of autoimmune encephalomyelitis (EAE), induced by immunization of rats, guinea pigs, or primates with CNS tissue myelin or recombinant MOG (Höftberger et al. 2020). Thus, EAE appears to be a very suitable model for MOGAD, but much less for MS. Support of this view is provided in a recent study, on archival material from a patient, who died in the 1950s with an acute MS-like inflammatory demyelinating disease after misguided repeated immunization with brain tissue (Höftberger et al. 2015). New molecular biology technologies allowed resurrection of a pathogenic auto-antibody from the archival formaldehyde fixed and paraffin embedded tissue, which was found to be directed against a conformational epitope of MOG and induced demyelination after transfer in vivo (Beltran et al. 2020). 4) Is subpial demyelination a unique feature of multiple sclerosis pathology? For a long time, MS has been regarded a demyelinating disease affecting the white matter. However, with the availability of highly sensitive immunocytochemical methods, which allow staining of the very thin myelinated fibers within the grey matter (Peterson et al. 2000), it became clear that demyelination in the grey matter in the MS brain is extensive (Kutzelnigg et al. 2005) and may even give rise to cortico-spinal or pure cortical variants of the disease in a subset of patients (Trapp et al. 2018). Three types of cortical lesions have been identified: the cortico-subcortical lesions (Type 1), the intracortical lesions (Type 2), and the subpial lesions (Type 3) (Bo et al. 2003). The third type is most abundant in MS and is related to focal inflammatory aggregates in the leptomeninges (Magliozzi et al. 2010; Figure 2). Several previous studies have indicated that subpial demyelinated lesions in the cortex may be specific for MS (Moll et al. 2008, Fischer et al. 2013), but this view was based on a rather limited sample of neuropathological conditions other than MS. This has now been changed in a very comprehensive study by Junker et al. (2020), in which nearly every thinkable neuropathological condition has been investigated for cortical demyelination. Interestingly, subpial demyelination was exclusively present in MS and not seen in any of the other conditions. The only key diagnosis missed in this case series was MOGAD. However, this was also specifically addressed in the systematic studies of MOGAD pathology outlined above (Höftberger et al. 2020, Takai et al. 2020), describing intracortical and cortico-subcortical lesions but absent subpial cortical lesions. In rare instances, these intracortical lesions can fuse and give rise to large focal confluent plaques of demyelination. Thus, so far it seems that subpial cortical demyelination is a unique feature of MS pathology, but the evidence for its absence in MOGAD is thus far based on a rather small sample of cases. Nevertheless, the presence of a subpial cortical lesion strongly supports a neuropathological diagnosis of MS.
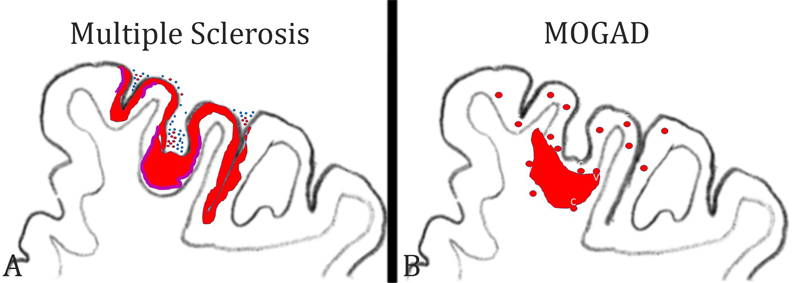
Figure 2: Patterns of cortical demyelination in MS and MOGAD: A) In MS, the dominant and most specific type of cortical demyelination is the subpial lesion. It is characterized by a band of demyelination spanning over several gyri and sulci (red lesions). The lesions are more extensive in the invaginations of the cortex. Active cortical demyelination is associated with meningeal inflammation by T-cells (blue dots) and B-cells (red dots) and is characterized by a band of activated macrophages / microglia at the border between the cortical lesion and the surrounding normal appearing cortex (purple bands). B) In MOGAD, the dominant cortical pathology is the presence of small perivenous intracortical demyelinated lesions (red circles). They arise around small cortical veins and venules with inflammatory infiltrates. On rare occasions, such intracortical perivenous lesions may give rise to a focal confluent cortical lesion, which frequently also expands into the adjacent subcortical white matter. 5) What immune cells drive inflammation in multiple sclerosis? Our understanding of the immune mechanisms driving inflammation in MS is largely biased by immunological studies of experimental autoimmune encephalomyelitis, which now turns out to have more relevance for MOGAD than for MS. Key mechanisms of inflammation, defined from such EAE studies, such as the key role of MHC Class II restricted T-cells responses, the involvement of CD4+ Th17 cells, or the central role of GM-CSF driven myeloid cell activation (Schreiner and Becher 2015) are not convincingly supported by recent therapeutic trials targeting the respective immunological pathways (Baker et al. 2017). In contrast, most effective therapeutic success is seen with drugs that target T-cells and B-cells together or even B-cells alone. Thus, it is still an unresolved question, as to what cells and immune mechanisms trigger or drive inflammation in MS. Pathology can help clarify this issue by providing a concise account of the types of inflammatory cells seen in different stages in the evolution of MS lesions. Until a few years ago, information about the composition of inflammatory infiltrates in MS was restricted to small studies performed on a limited number of patients and lesions. Since 2017, however, there are now three large studies available, which performed a phenotyping of inflammatory cells in an overall sample of more than 200 MS patients and which included all stages of the disease (Van Nierop et al. 2017, Machado Santos et al. 2018, Fransen et al. 2020). All studies reached a similar conclusion that the dominant lymphocytes in the MS lesions are tissue resident CD8+ effector memory T-cells (TTRM), while infiltration with CD4+ T-cells is sparse. This is the case in all types of MS, including fulminant acute MS, relapsing remitting MS as well as primary or secondary progressive MS and is the same in all activity stages of the lesions. In active lesions, a subset of CD8+ cells show focally and temporally restricted activation, and B-cells are much more numerous in early stages of the disease and in active lesions than in inactive lesions at late stages (Machado Santos et al. 2018). In a recent study, imaging mass cytometry was applied to the biopsy tissue of a single MS patient who suffered from a rebound exacerbation after cessation of natalizumab treatment (Ramaglia et al. 2019). Also, in this study, the infiltrates contained T-cells and B-cells, but the relative proportion of CD4+ T-cells was higher compared to that seen in the previous studies. However, activation of T-cells, visualized by the expression of the transcription factor NFAT or proliferation markers, was seen only in CD8+ T-cells. Besides their absolute numbers, the activation state of leukocytes is relevant for driving the inflammatory reaction. Similarly as before (Machado Santos et al. 2018), the most frequent B-cell phenotypes were IgG+ CD38+ plasmablasts. Thus, overall, the pathological data are in line with the observed effects of anti-inflammatory treatments in MS patients. 6) What do the inflammatory cells in MS lesions recognize? An important open question is what antigen is recognized by the CD8+ tissue resident memory T-cells. Since such T-cells acquire their phenotype and persist after effective clearance of their cognate antigen and become reactivated when the antigen re-appears, it is unlikely that these cells recognize a classical auto-antigen, which is present in the CNS in excess all the time. In line with this view, no reactivity of CD8+ T-cells from MS brain lesions has been seen against the commonly tested myelin antigens (van Nierop et al. 2017). Several potential candidate antigens have recently been identified. Since accumulating epidemiological evidence associates Epstein Barr Virus infection with MS (Bar-Or et al. 2020, Levin et al. 2010), one hypothesis is that B-cells with latent EBV infection are present within the CNS of MS patients (Serafini et al. 2007, Veroni et al. 2018) together with EBV-reactive CD8+ tissue resident effector memory T-cells (Serafini et al. 2020). Here, the hypothesis is that inflammatory activity is triggered and propagated when the virus is activated in latently infected cells and recognized by the specific T-cells (Serafini et al. 2020). As a note of caution, several other groups have tried to confirm the specific presence of EBV infected B-cells in the MS brain and lesions and have failed (Lassmann et al. 2010, van Nierop et al. 2017). The reason for these discrepancies is still unresolved. In a similar approach, an interaction between brain resident CD8+ cells and B-cells has been described, where the B-cells did not express EBV (van Nierop et al. 2017). This suggests that the cells recognize a B-cell autoantigen. A possible mechanism for how such autoimmunity against B-cells may be induced is provided by Jelcic et al. (2018). MS B-cells, possibly in relation to their EBV infection status, expand by auto-proliferation and present a B-cell specific auto-antigen to CD4+ T-cells, which also expand and infiltrate the brain and spinal cord. Within the CNS, these T-cells are activated by antigen recognition on infiltrating B-cells. In addition, the respective auto-antigen (RASGRP2) is also expressed in neurons, which may further propagate inflammation and tissue injury. Whether this mechanism also accounts for activation of CD8+ T-cells, the dominant inflammatory cells in MS lesions, is unknown. A different approach has been followed by Konjevic Sabolek et al. (2019). In this study, the interaction of CD8+ T-cells with local target cells was identified by the polarized location of cytotoxic granules at the site of contact. When this was seen, the target cell was isolated and its nature was determined in gene expression studies and by immunohistochemistry. In this study, the only cells which interacted with cytotoxic CD8+ T-cells in MS lesions were myeloid cells, expressing markers of macrophages and microglia, suggesting these may harbor the target antigen(s). Finally, other studies suggest that the target antigen recognized by T-cells in MS lesions may be the stress protein alpha B crystallin, which is in MS lesions most prominently expressed in active lesions (van Noort et al. 2010). In summary, the new data from systematic phenotypical and functional studies on the inflammatory response within MS lesions provide groundbreaking new insights into the pathophysiology of the disease and question the concept of MS being an autoimmune disease against myelin. However, the results are in part very divergent and, to some extent, contradicting. Whether this indicates heterogeneous disease mechanisms between individual patients or patient subgroups will be seen in the future. 7) Which microglia or macrophage phenotypes are associated with tissue damage in the brain? Microglia and recruited macrophages play a major role in the induction of tissue injury, not only in inflammatory brain lesions but also in neurodegenerative diseases. For a long time, it was difficult to distinguish between activated microglia and recruited macrophages within brain lesions. This has changed with the introduction of markers, which are selectively expressed on microglia, such as the membrane protein TMEM 119 and the marker for homeostatic microglia P2RY12. These markers have been used in a number of studies on various different inflammatory and neurodegenerative conditions and revealed a surprisingly uniform reaction pattern of myeloid cells in human diseases (Zrzavy et al. 2017, 2018, Hayashida et al. 2020, Jäckle et al. 2020). In essence, at sites of initial tissue injury, resident microglia become activated in a pro-inflammatory pattern. This initial step is followed by recruitment of myeloid cells from the circulation, and the recruited cells also show pro-inflammatory activation or, at later stages of lesion maturation, an intermediate phenotype. The distinction of pro- versus anti-inflammatory phenotypes is now seen in a much more critical way, since some pro-inflammatory actions, such as the interaction of macrophages with target cells may contribute to tissue damage but may also be essential for neuroprotection and regeneration through the clearance of debris (Cignarella et al. 2020). An important pro-inflammatory type of activation, which seems to play a major role in the induction of tissue injury, is the expression of proteins involved in oxidative stress, such as the expression of the NOX2 complex (NADPH oxidase complex 2), a prominent marker of microglia expressed in active lesions in different inflammatory conditions as well as vascular or neurodegenerative diseases in humans (Zrzavy et al. 2017, Fischer et al. 2013). To define the patterns of microglia activation in such diseases may finally result in more selective and efficient neuroprotective treatments. This is now possible with new technologies, such as single-cell RNA sequencing or immunocytochemical methods, which allow the simultaneous detection of multiple protein antigens within the same section, such as imaging mass cytometry. In some studies of models of autoimmune encephalomyelitis and MS, these techniques have been used and confirmed in an elegant way the patterns of microglia activation and macrophage recruitment, which have been described before with more conventional techniques as summarized above (Masuda et al. 2019, Ramaglia et al. 2020). However, they additionally showed that microglia and macrophages with different phenotypes are present side-by-side in the same lesion. This may indicate selective activation signals in specific subpopulations of cells. Not surprisingly, the data showed that different MS lesions, which were in the same activity stage but still displayed some distinct morphological features, were infiltrated by different subsets of myeloid cells (Masuda et al. 2019). However, these studies did not reveal the high expression of molecules involved in oxidative injury, which had been prominent in earlier more conventional neuropathological studies. A possible explanation for these discrepancies is provided by an elegant study, which specifically focused on microglia activation and oxidative injury in autoimmune encephalomyelitis (Mendiola et al. 2020). The authors first defined the molecular phenotype of microglia specifically selected from areas of oxidative injury. Incorporating this profile in the analysis of single-cell RNA sequencing data and immunocytochemistry, they identified specific microglia and macrophage subpopulations, which trigger oxidative stress, and showed that these are selective subtypes of cells within the population of activated myeloid cells. These particular cell types also express molecules that are important in the coagulation cascade and in glutathione metabolism. In addition, they produce molecules, which counteract oxidative stress. One of these molecules Acivivin, a glutathione regulating compound, suppresses the inflammatory tissue damage in a model of autoimmune encephalomyelitis. It will be instrumental in the future to validate directly the relevance of these findings in MS lesions and also in vascular or neurodegenerative diseases of the central nervous system. The presence of a subtype of myeloid cells triggering and resolving oxidative stress could reconcile the above discrepancies and provide a mechanism for the excessive oxidative injury in the lesions and the associations of markers for oxidative injury with markers for disturbed blood coagulation in the lesions. In the long run, these data may lead to tools for the therapeutic blockade of one of the most detrimental aspects of microglia activation in human brain disease. 8) How do vascular comorbidities influence multiple sclerosis patients? Clinical epidemiology of MS has shown that patients with vascular risk factors, such as diabetes, hypercholesterolaemia, hypertension, or heart disease have a more aggressive disability progression in comparison to patients lacking these co-morbidities (Marrie et al. 2010). This is also reflected by lower brain volumes (Pichler et al. 2019). Furthermore, persistence of inflammatory demyelinating lesions and higher lesion volumes are present in brain areas with a blood perfusion that is lower than in other brain areas (Haider et al. 2016). Such a synergism in neurodegeneration may in part be explained by shared effector mechanisms of tissue injury between vascular and inflammatory diseases, including microglia activation, oxidative injury, and mitochondrial damage (Zrzavy et al. 2017, 2018, Mahad et al. 2015). A strategically important piece in the puzzle of vascular comorbidities and MS, which was missing so far, was the lack of a comprehensive neuropathological description of systemic and intracranial vascular pathology in patients versus controls. This information has now been provided by a study, which is based on a unique archival collection of MS and control autopsy cases, where detailed information regarding systemic vascular diseases was recorded, and which was collected prior to the availability of disease modifying treatments (Geraldes et al. 2020). The study shows, as expected, that systemic and intracranial vascular abnormalities increase with age in both the MS and the control group. Young MS patients appear to have a moderate reduction of systemic vascular co-morbidities compared to controls, but this difference disappears with aging. Within the central nervous system, MS patients showed a profound increase in small arteries with increased perivascular space, perivascular hemosiderin depositions and periarteriolar accumulation of inflammatory cells, a type of pathology which correlated in its extent with the degree of MS-related pathology. These results underline the presence of age-related vascular co-morbidities in the brain of MS patients, which may be an amplification factor for neurodegeneration and disease progression in aging patients. Additionally, they indicate that, in contrast to the current view, vascular pathology is not restricted to veins but also affects small arteries, and this arterial pathology develops at least in part independently from systemic vascular risk factors. In particular, the mechanisms how inflammatory infiltrates accumulate around arteries in the MS brain requires further attention. 9) What is wrong with remyelination in multiple sclerosis? Primary demyelination with preservation of axons is the pathological hallmark of inflammatory demyelinating diseases, such as MS. Myelin allows saltatory conduction in axons and also protects the axons from neurodegeneration. Thus, major efforts in MS research have been devoted to develop treatments which prevent demyelination or stimulate myelin repair (Lubetzki et al. 2020). These repair strategies, mainly designed to provide oligodendrocyte progenitor cells in the lesions and to stimulate their differentiation into myelinating oligodendrocytes, have been developed and tested in experimental models of demyelinating disease and found to be quite effective. However, none of the strategies have yet resulted in clinically meaningful myelin repair in controlled clinical trials in MS patients. In experimental models of demyelinating disease, profound spontaneous remyelination is the rule, and remyelination stimulating therapies in essence accelerate myelin repair. Although spontaneous remyelination may also occur in MS patients, in particular in fresh lesions at early disease stages, remyelination is, in general, sparse or absent. Several different mechanisms have been suggested to be responsible for this remyelination failure in MS lesions. It may be due to a genuine defect in oligodendrocyte (progenitor) biology. In an elegant study, Starost et al. (2020) approached this question by studying oligodendrocytes, induced from pluripotent stem cells from MS patients and controls. They show that there is no difference in the proliferation, differentiation and myelin production between such cells derived from MS patients and from controls, thus strongly suggesting that there is no genuine defect of oligodendrocytes in MS patients, though with the caveat that this was only performed in cells induced to form oligodendrocytes with expression of transcriptional factors from three MS patients and three controls. In a parallel neuropathological approach, the same group analyzed the patterns or remyelination in different disease stages of MS and in different stages of lesion formation (Heß et al. 2020). They found that remyelination is highly efficient in a subset of active lesions in the early stage of the disease, and their data suggest that the remyelinating cells may be derived from mature oligodendrocytes that had survived active myelin destruction. This has also been suggested in recent studies, using radiocarbon methods to determine the time of birth of new oligodendrocytes in MS lesions (Yeung et al. 2019). In contrast, in chronic active lesions at later stages of MS, very little remyelination was seen and the lack of remyelination was associated with pro-inflammatory activity of the local microglia population (Heß et al. 2020). These data suggest that products of activated inflammatory cells in chronic MS lesions may not only lead to their slow expansion but also inhibit the repair of myelin. This finding is also supported by the other study (Starost et al. 2020), which provides convincing evidence that pro-inflammatory mediators, in particular gamma-interferon, blocks the differentiation of inducible pluripotent stem cells into myelinating oligodendrocytes. The key importance of these studies is that they document that the process of remyelination failure in MS is complex and not fully reflected in the current experimental models of inflammatory demyelinating disease. Thus, for test screening of remyelination-promoting therapies different systems have to be used, which reproduce the inflammation-associated remyelination failure. 10) What can we learn from Down Syndrome about inflammation and neurodegeneration in Alzheimer’s disease? A large spectrum of data coming from experimental studies as well as from neuropathological and genetic investigations suggest an important role of innate immune mechanisms in the pathogenesis of Alzheimer’s disease (Akiyama et al. 2000). In particular, microglia phenotype and function are associated with progression of cognitive decline. However, information regarding the time course and type of microglia pathology especially in the pre-symptomatic phases of the disease is limited. Analyzing Down syndrome brain pathology of patients may provide some answers to these questions, since a gene dosage effect on amyloid precursor protein production predictably results in early onset Alzheimer’s disease (Wisniewski et al. 1985, Ballard et al. 2016). This approach was followed in a recent study, in which the patterns of microglia activation and the expression of pro- and anti-inflammatory cytokines was analyzed in a large sample of brain tissue of patients with trisomy 21 who had died at different ages (Flores Aguilar et al. 2020). Already, in juvenile patients, a long time before the first accumulation of soluble Aß or the first deposition of Aß-plaques, microglia activation and the production of proinflammatory cytokines was elevated in comparison to age matched controls. In contrast, in older patients, when initial Alzheimer’s disease pathology became apparent, microglia activation decreased and was replaced by senescent microglia phenotypes as well as a reduction of the production of pro-inflammatory cytokines. These results further support the view that anti-inflammatory treatment strategies in Alzheimer’s disease may be most beneficial when applied in very early (pre-symptomatic) stages of disease evolution. The data further suggest that the inclusion of patients with trisomy 21 in future clinical trials could provide a valuable contribution to improve treatment options in patients with Alzheimer’s disease. Conclusions Neuroimmunology is a highly dynamic field providing fascinating new insights into the pathogenesis of brain inflammation and neurodegeneration. The bulk of data, however, describe experimental models and, thus, an additional step is necessary, which defines the relevance of the data for human disease. For this, neuropathological studies are essential, but they have to be based on systematic analysis of a broad spectrum of cases and controls and have to be performed with the most suitable molecular or immunological technology. In this short review the prime focus was laid on such studies of human disease, which were sufficiently powered to provide definite answers to burning questions of neuroinflammation and inflammatory brain diseases. References Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al.. Inflammation and Alzheimer’s disease. Neurobiol Aging 2000; 21: 383–421. doi: 10.1016/s0197-4580(00)00124-x. Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K. Memory B Cells are Major Targets for Effective Immunotherapy in Relapsing Multiple Sclerosis. EBioMedicine. 2017 Feb;16:41-50. doi: 10.1016/j.ebiom.2017.01.042. Epub 2017 Jan 31. PMID: 28161400; PMCID: PMC5474520. Ballard C, Mobley W, Hardy J, Williams G, Corbett A. Dementia in Down’s syndrome. Lancet Neurol 2016; 15: 622–36. doi: 10.1016/S1474-4422(16)00063-6. Epub 2016 Apr 11. Bar-Or A, Pender MP, Khanna R, Steinman L, Hartung HP, Maniar T, Croze E, Aftab BT, Giovannoni G, Joshi MA. Epstein-Barr Virus in Multiple Sclerosis: Theory and Emerging Immunotherapies. Trends Mol Med. 2020 Mar;26(3):296-310. doi: 10.1016/j.molmed.2019.11.003. Epub 2019 Dec 17. PMID: 31862243; PMCID: PMC7106557. Beltrán E, Paunovic M, Gebert D, Cesur E, Jeitler M, Höftberger R, Malotka J, Mader S, Kawakami N, Meinl E, Bradl M, Dornmair K, Lassmann H. Archeological neuroimmunology: resurrection of a pathogenic immune response from a historical case sheds light on human autoimmune encephalomyelitis and multiple sclerosis. Acta Neuropathol. 2020 Oct 29. doi: 10.1007/s00401-020-02239-2. Epub ahead of print. PMID: 33242149. Bø L, Vedeler CA, Nyland HI, Trapp BD, Mørk SJ. Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol. 2003 Jul;62(7):723-32. doi: 10.1093/jnen/62.7.723. PMID: 12901699. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T, Anastasina M, Smura T, Levanov L, Szirovicza L, Tobi A, Kallio-Kokko H, Österlund P, Joensuu M, Meunier FA, Butcher SJ, Winkler MS, Mollenhauer B, Helenius A, Gokce O, Teesalu T, Hepojoki J, Vapalahti O, Stadelmann C, Balistreri G, Simons M. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020 Nov 13;370(6518):856-860. doi: 10.1126/science.abd2985. Epub 2020 Oct 20. PMID: 33082293. Carceles-Cordon M, Mannara F, Aguilar E, Castellanos A, Planagumà J, Dalmau J. NMDAR Antibodies Alter Dopamine Receptors and Cause Psychotic Behavior in Mice. Ann Neurol. 2020 Sep;88(3):603-613. doi: 10.1002/ana.25829. Epub 2020 Jul 11. PMID: 32583480. Cignarella F, Filipello F, Bollman B, Cantoni C, Locca A, Mikesell R, Manis M, Ibrahim A, Deng L, Benitez BA, Cruchaga C, Licastro D, Mihindukulasuriya K, Harari O, Buckland M, Holtzman DM, Rosenthal A, Schwabe T, Tassi I, Piccio L. TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 2020 Oct;140(4):513-534. doi: 10.1007/s00401-020-02193-z. Epub 2020 Aug 9. PMID: 32772264; PMCID: PMC7498497. Deigendesch N, Sironi L, Kutza M, Wischnewski S, Fuchs V, Hench J, Frank A, Nienhold R, Mertz KD, Cathomas G, Matter MS, Siegemund M, Tolnay M, Schirmer L, Pröbstel AK, Tzankov A, Frank S. Correlates of critical illness-related encephalopathy predominate postmortem COVID-19 neuropathology. Acta Neuropathol. 2020 Oct;140(4):583-586. doi: 10.1007/s00401-020-02213-y. Epub 2020 Aug 26. PMID: 32851506; PMCID: PMC7449525. Fischer MT, Wimmer I, Höftberger R, Gerlach S, Haider L, Zrzavy T, Hametner S, Mahad D, Binder CJ, Krumbholz M, Bauer J, Bradl M, Lassmann H. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013 Jun;136(Pt 6):1799-815. doi: 10.1093/brain/awt110. Epub 2013 May 17. PMID: 23687122; PMCID: PMC3673462. Flores-Aguilar L, Iulita MF, Kovecses O, Torres MD, Levi SM, Zhang Y, Askenazi M, Wisniewski T, Busciglio J, Cuello AC. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain. 2020 Nov 18:awaa326. doi: 10.1093/brain/awaa326. Epub ahead of print. PMID: 33206953. Fransen NL, Hsiao CC, van der Poel M, Engelenburg HJ, Verdaasdonk K, Vincenten MCJ, Remmerswaal EBM, Kuhlmann T, Mason MRJ, Hamann J, Smolders J, Huitinga I. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain. 2020 Jun 1;143(6):1714-1730. doi: 10.1093/brain/awaa117. PMID: 32400866. Fujihara K, Cook LJ. Neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody-associated disease: current topics. Curr Opin Neurol. 2020 Jun;33(3):300-308. doi: 10.1097/WCO.0000000000000828. PMID: 32374571. Geraldes R, Esiri MM, Perera R, Yee SA, Jenkins D, Palace J, DeLuca GC. Vascular disease and multiple sclerosis: a post-mortem study exploring their relationships. Brain. 2020 Oct 1;143(10):2998-3012. doi: 10.1093/brain/awaa255. PMID: 32875311. Haider L, Zrzavy T, Hametner S, Höftberger R, Bagnato F, Grabner G, Trattnig S, Pfeifenbring S, Brück W, Lassmann H. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain. 2016 Mar;139(Pt 3):807-15. doi: 10.1093/brain/awv398. Epub 2016 Feb 8. PMID: 26912645; PMCID: PMC4766379. Hayashida S, Masaki K, Suzuki SO, Yamasaki R, Watanabe M, Koyama S, Isobe N, Matsushita T, Takahashi K, Tabira T, Iwaki T, Kira JI. Distinct microglial and macrophage distribution patterns in the concentric and lamellar lesions in Baló's disease and neuromyelitis optica spectrum disorders. Brain Pathol. 2020 Sep 9. doi: 10.1111/bpa.12898. Epub ahead of print. PMID: 32902014. Hernández-Fernández F, Sandoval Valencia H, Barbella-Aponte RA, Collado-Jiménez R, Ayo-Martín Ó, Barrena C, Molina-Nuevo JD, García-García J, Lozano-Setién E, Alcahut-Rodriguez C, Martínez-Martín Á, Sánchez-López A, Segura T. Cerebrovascular disease in patients with COVID-19: neuroimaging, histological and clinical description. Brain. 2020 Oct 1;143(10):3089-3103. doi: 10.1093/brain/awaa239. PMID: 32645151; PMCID: PMC7454411. Heß K, Starost L, Kieran NW, Thomas C, Vincenten MCJ, Antel J, Martino G, Huitinga I, Healy L, Kuhlmann T. Lesion stage-dependent causes for impaired remyelination in MS. Acta Neuropathol. 2020 Sep;140(3):359-375. doi: 10.1007/s00401-020-02189-9. Epub 2020 Jul 24. PMID: 32710244; PMCID: PMC7424408. Höftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, Hochmeister S, Joldic D, Pittock SJ, Tillema JM, Gorman M, Lassmann H, Lucchinetti CF. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020 May;139(5):875-892. doi: 10.1007/s00401-020-02132-y. Epub 2020 Feb 11. PMID: 32048003; PMCID: PMC7181560. Höftberger R, Lassmann H. Immune-mediated disorders. Handb Clin Neurol. 2017;145:285-299. doi: 10.1016/B978-0-12-802395-2.00020-1. PMID: 28987176. Höftberger R, Leisser M, Bauer J, Lassmann H. Autoimmune encephalitis in humans: how closely does it reflect multiple sclerosis ? Acta Neuropathol Commun. 2015 Dec 4;3:80. doi: 10.1186/s40478-015-0260-9. PMID: 26637427; PMCID: PMC4670499. Jäckle K, Zeis T, Schaeren-Wiemers N, Junker A, van der Meer F, Kramann N, Stadelmann C, Brück W. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain. 2020 Jul 1;143(7):2073-2088. doi: 10.1093/brain/awaa158. PMID: 32577755. Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, Madjovski A, Ruhrmann S, Faigle W, Frauenknecht K, Pinilla C, Santos R, Hammer C, Ortiz Y, Opitz L, Grönlund H, Rogler G, Boyman O, Reynolds R, Lutterotti A, Khademi M, Olsson T, Piehl F, Sospedra M, Martin R. Memory B Cells Activate Brain-Homing, Autoreactive CD4+ T Cells in Multiple Sclerosis. Cell. 2018 Sep 20;175(1):85-100.e23. doi: 10.1016/j.cell.2018.08.011. Epub 2018 Aug 30. PMID: 30173916; PMCID: PMC6191934. Jensen MP, Le Quesne J, Officer-Jones L, Teodòsio A, Thaventhiran J, Ficken C, Goddard M, Smith C, Menon D, Allinson KSJ. Neuropathological findings in two patients with fatal COVID-19. Neuropathol Appl Neurobiol. 2020 Sep 8. doi: 10.1111/nan.12662. Epub ahead of print. PMID: 32895961. Junker A, Wozniak J, Voigt D, Scheidt U, Antel J, Wegner C, Brück W, Stadelmann C. Extensive subpial cortical demyelination is specific to multiple sclerosis. Brain Pathol. 2020 May;30(3):641-652. doi: 10.1111/bpa.12813. Epub 2020 Feb 3. PMID: 31916298. Kabbani N, Olds JL. Does COVID19 Infect the Brain? If So, Smokers Might Be at a Higher Risk. Mol Pharmacol. 2020 May;97(5):351-353. doi: 10.1124/molpharm.120.000014. Epub 2020 Apr 1. PMID: 32238438; PMCID: PMC7237865. Kantonen J, Mahzabin S, Mäyränpää MI, Tynninen O, Paetau A, Andersson N, Sajantila A, Vapalahti O, Carpén O, Kekäläinen E, Kantele A, Myllykangas L. Neuropathologic features of four autopsied COVID-19 patients. Brain Pathol. 2020 Aug 6:10.1111/bpa.12889. doi: 10.1111/bpa.12889. Epub ahead of print. PMID: 32762083; PMCID: PMC7436498. Kanwar D, Baig AM, Wasay M. Neurological manifestations of COVID-19. J Pak Med Assoc. 2020 May;70(Suppl 3)(5):S101-S103. doi: 10.5455/JPMA.20. PMID: 32515379. Konjevic Sabolek M, Held K, Beltrán E, Niedl AG, Meinl E, Hohlfeld R, Lassmann H, Dornmair K. Communication of CD8+ T cells with mononuclear phagocytes in multiple sclerosis. Ann Clin Transl Neurol. 2019 Jul;6(7):1151-1164. doi: 10.1002/acn3.783. Epub 2019 Jun 14. PMID: 31353869; PMCID: PMC6649540. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Brück W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005 Nov;128(Pt 11):2705-12. doi: 10.1093/brain/awh641. Epub 2005 Oct 17. PMID: 16230320. Landa J, Gaig C, Plagumà J, Saiz A, Antonell A, Sanchez-Valle R, Dalmau J, Graus F, Sabater L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann Neurol. 2020 Nov;88(5):1023-1027. doi: 10.1002/ana.25857. Epub 2020 Aug 27. PMID: 32740999. Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM; NeuroproMiSe EBV Working Group. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue--report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. 2011 Sep;134(Pt 9):2772-86. doi: 10.1093/brain/awr197. Epub 2011 Aug 16. PMID: 21846731; PMCID: PMC3170536. Levin LI, Munger KL, O'Reilly EJ, Falk KI, Ascherio A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol. 2010 Jun;67(6):824-30. doi: 10.1002/ana.21978. PMID: 20517945; PMCID: PMC3089959. Li YC, Bai WZ, Hashikawa T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol. 2020 Jun;92(6):552-555. doi: 10.1002/jmv.25728. Epub 2020 Mar 11. PMID: 32104915; PMCID: PMC7228394. Liu JM, Tan BH, Wu S, Gui Y, Suo JL, Li YC. Evidence of central nervous system infection and neuroinvasive routes, as well as neurological involvement, in the lethality of SARS-CoV-2 infection. J Med Virol. 2020 Oct 1:10.1002/jmv.26570. doi: 10.1002/jmv.26570. Epub ahead of print. PMID: 33002209; PMCID: PMC7537172. Lubetzki C, Zalc B, Williams A, Stadelmann C, Stankoff B. Remyelination in multiple sclerosis: from basic science to clinical translation. Lancet Neurol. 2020 Aug;19(8):678-688. doi: 10.1016/S1474-4422(20)30140-X. PMID: 32702337. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, Gabriely G, Bien CG, Bauer J, Lassmann H. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. 2018 Jul 1;141(7):2066-2082. doi: 10.1093/brain/awy151. PMID: 29873694; PMCID: PMC6022681. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, Aloisi F, Reynolds R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010 Oct;68(4):477-93. doi: 10.1002/ana.22230. PMID: 20976767. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015 Feb;14(2):183-93. doi: 10.1016/S1474-4422(14)70256-X. PMID: 25772897. Marrie RA, Rudick R, Horwitz R, Cutter G, Tyry T, Campagnolo D, Vollmer T. Vascular comorbidity is associated with more rapid disability progression in multiple sclerosis. Neurology. 2010 Mar 30;74(13):1041-7. doi: 10.1212/WNL.0b013e3181d6b125. PMID: 20350978; PMCID: PMC2848107. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, Coenen VA, Reinacher PC, Michel A, Sure U, Gold R, Grün D, Priller J, Stadelmann C, Prinz M. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019 Feb;566(7744):388-392. doi: 10.1038/s41586-019-0924-x. Epub 2019 Feb 13. Erratum in: Nature. 2019 Apr;568(7751):E4. PMID: 30760929. Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C, Mushumba H, Fitzek A, Allweiss L, Dandri M, Dottermusch M, Heinemann A, Pfefferle S, Schwabenland M, Sumner Magruder D, Bonn S, Prinz M, Gerloff C, Püschel K, Krasemann S, Aepfelbacher M, Glatzel M. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 2020 Nov;19(11):919-929. doi: 10.1016/S1474-4422(20)30308-2. Epub 2020 Oct 5. PMID: 33031735; PMCID: PMC7535629. Matute C, Palma A, Serrano-Regal MP, Maudes E, Barman S, Sánchez-Gómez MV, Domercq M, Goebels N, Dalmau J. N-Methyl-D-Aspartate Receptor Antibodies in Autoimmune Encephalopathy Alter Oligodendrocyte Function. Ann Neurol. 2020 May;87(5):670-676. doi: 10.1002/ana.25699. Epub 2020 Feb 24. PMID: 32052483. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, Laue M, Schneider J, Brünink S, Greuel S, Lehmann M, Hassan O, Aschman T, Schumann E, Chua RL, Conrad C, Eils R, Stenzel W, Windgassen M, Rößler L, Goebel HH, Gelderblom HR, Martin H, Nitsche A, Schulz-Schaeffer WJ, Hakroush S, Winkler MS, Tampe B, Scheibe F, Körtvélyessy P, Reinhold D, Siegmund B, Kühl AA, Elezkurtaj S, Horst D, Oesterhelweg L, Tsokos M, Ingold-Heppner B, Stadelmann C, Drosten C, Corman VM, Radbruch H, Heppner FL. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. 2020 Nov 30. doi: 10.1038/s41593-020-00758-5. Epub ahead of print. PMID: 33257876. Mendiola AS, Ryu JK, Bardehle S, Meyer-Franke A, Ang KK, Wilson C, Baeten KM, Hanspers K, Merlini M, Thomas S, Petersen MA, Williams A, Thomas R, Rafalski VA, Meza-Acevedo R, Tognatta R, Yan Z, Pfaff SJ, Machado MR, Bedard C, Rios Coronado PE, Jiang X, Wang J, Pleiss MA, Green AJ, Zamvil SS, Pico AR, Bruneau BG, Arkin MR, Akassoglou K. Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat Immunol. 2020 May;21(5):513-524. doi: 10.1038/s41590-020-0654-0. Epub 2020 Apr 13. Erratum in: Nat Immunol. 2020 Sep;21(9):1135. PMID: 32284594; PMCID: PMC7523413. Moll NM, Rietsch AM, Ransohoff AJ, Cossoy MB, Huang D, Eichler FS, Trapp BD, Ransohoff RM. Cortical demyelination in PML and MS: Similarities and differences. Neurology. 2008 Jan 29;70(5):336-43. doi: 10.1212/01.WNL.0000284601.54436.e4. PMID: 17914063. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, Jayaseelan DL, Kumar G, Raftopoulos RE, Zambreanu L, Vivekanandam V, Khoo A, Geraldes R, Chinthapalli K, Boyd E, Tuzlali H, Price G, Christofi G, Morrow J, McNamara P, McLoughlin B, Lim ST, Mehta PR, Levee V, Keddie S, Yong W, Trip SA, Foulkes AJM, Hotton G, Miller TD, Everitt AD, Carswell C, Davies NWS, Yoong M, Attwell D, Sreedharan J, Silber E, Schott JM, Chandratheva A, Perry RJ, Simister R, Checkley A, Longley N, Farmer SF, Carletti F, Houlihan C, Thom M, Lunn MP, Spillane J, Howard R, Vincent A, Werring DJ, Hoskote C, Jäger HR, Manji H, Zandi MS. The emerging spectrum of COVID-19 neurology: clinical, radiological and laboratory findings. Brain. 2020 Oct 1;143(10):3104-3120. doi: 10.1093/brain/awaa240. PMID: 32637987; PMCID: PMC7454352. Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001 Sep;50(3):389-400. doi: 10.1002/ana.1123. PMID: 11558796. Pichler A, Khalil M, Langkammer C, Pinter D, Ropele S, Fuchs S, Bachmaier G, Enzinger C, Fazekas F. The impact of vascular risk factors on brain volume and lesion load in patients with early multiple sclerosis. Mult Scler. 2019 Jan;25(1):48-54. doi: 10.1177/1352458517736149. Epub 2017 Oct 13. PMID: 29027843. Pitsch J, Kamalizade D, Braun A, Kuehn JC, Gulakova PE, Rüber T, Lubec G, Dietrich D, von Wrede R, Helmstaedter C, Surges R, Elger CE, Hattingen E, Vatter H, Schoch S, Becker AJ. Drebrin Autoantibodies in Patients with Seizures and Suspected Encephalitis. Ann Neurol. 2020 Jun;87(6):869-884. doi: 10.1002/ana.25720. Epub 2020 Apr 10. PMID: 32196746. Ramaglia V, Sheikh-Mohamed S, Legg K, Park C, Rojas OL, Zandee S, Fu F, Ornatsky O, Swanson EC, Pitt D, Prat A, McKee TD, Gommerman JL. Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. Elife. 2019 Aug 1;8:e48051. doi: 10.7554/eLife.48051. PMID: 31368890; PMCID: PMC6707785. Rauschenberger V, von Wardenburg N, Schaefer N, Ogino K, Hirata H, Lillesaar C, Kluck CJ, Meinck HM, Borrmann M, Weishaupt A, Doppler K, Wickel J, Geis C, Sommer C, Villmann C. Glycine Receptor Autoantibodies Impair Receptor Function and Induce Motor Dysfunction. Ann Neurol. 2020 Sep;88(3):544-561. doi: 10.1002/ana.25832. Epub 2020 Jul 20. PMID: 32588476. Reichard RR, Kashani KB, Boire NA, Constantopoulos E, Guo Y, Lucchinetti CF. Neuropathology of COVID-19: a spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology. Acta Neuropathol. 2020 Jul;140(1):1-6. doi: 10.1007/s00401-020-02166-2. Epub 2020 May 24. PMID: 32449057; PMCID: PMC7245994. Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019 Feb;15(2):89-102. doi: 10.1038/s41582-018-0112-x. PMID: 30559466. Schreiner B, Becher B. Perspectives on cytokine-directed therapies in multiple sclerosis. Swiss Med Wkly. 2015 Oct 23;145:w14199. doi: 10.4414/smw.2015.14199. PMID: 26495801. Sechi E, Markovic SN, McKeon A, Dubey D, Liewluck T, Lennon VA, Lopez-Chiriboga AS, Klein CJ, Mauermann M, Pittock SJ, Flanagan EP, Zekeridou A. Neurologic autoimmunity and immune checkpoint inhibitors: Autoantibody profiles and outcomes. Neurology. 2020 Oct 27;95(17):e2442-e2452. doi: 10.1212/WNL.0000000000010632. Epub 2020 Aug 13. PMID: 32796130; PMCID: PMC7682911. Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, Andreoni L, Trivedi P, Salvetti M, Faggioni A, Aloisi F. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007 Nov 26;204(12):2899-912. doi: 10.1084/jem.20071030. Epub 2007 Nov 5. PMID: 17984305; PMCID: PMC2118531. Serafini B, Rosicarelli B, Veroni C, Mazzola GA, Aloisi F. Epstein-Barr Virus-Specific CD8 T Cells Selectively Infiltrate the Brain in Multiple Sclerosis and Interact Locally with Virus-Infected Cells: Clue for a Virus-Driven Immunopathological Mechanism. J Virol. 2019 Nov 26;93(24):e00980-19. doi: 10.1128/JVI.00980-19. PMID: 31578295; PMCID: PMC6880158. Starost L, Lindner M, Herold M, Xu YKT, Drexler HCA, Heß K, Ehrlich M, Ottoboni L, Ruffini F, Stehling M, Röpke A, Thomas C, Schöler HR, Antel J, Winkler J, Martino G, Klotz L, Kuhlmann T. Extrinsic immune cell-derived, but not intrinsic oligodendroglial factors contribute to oligodendroglial differentiation block in multiple sclerosis. Acta Neuropathol. 2020 Nov;140(5):715-736. doi: 10.1007/s00401-020-02217-8. Epub 2020 Sep 7. PMID: 32894330; PMCID: PMC7547031. Takai Y, Misu T, Kaneko K, Chihara N, Narikawa K, Tsuchida S, Nishida H, Komori T, Seki M, Komatsu T, Nakamagoe K, Ikeda T, Yoshida M, Takahashi T, Ono H, Nishiyama S, Kuroda H, Nakashima I, Suzuki H, Bradl M, Lassmann H, Fujihara K, Aoki M; Japan MOG-antibody Disease Consortium. Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain. 2020 May 1;143(5):1431-1446. doi: 10.1093/brain/awaa102. PMID: 32412053. Trapp BD, Vignos M, Dudman J, Chang A, Fisher E, Staugaitis SM, Battapady H, Mork S, Ontaneda D, Jones SE, Fox RJ, Chen J, Nakamura K, Rudick RA. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: a retrospective study. Lancet Neurol. 2018 Oct;17(10):870-884. doi: 10.1016/S1474-4422(18)30245-X. Epub 2018 Aug 22. PMID: 30143361; PMCID: PMC6197820. van Nierop GP, van Luijn MM, Michels SS, Melief MJ, Janssen M, Langerak AW, Ouwendijk WJD, Hintzen RQ, Verjans GMGM. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. 2017 Sep;134(3):383-401. doi: 10.1007/s00401-017-1744-4. Epub 2017 Jun 17. PMID: 28624961; PMCID: PMC5563341. van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L, Amor S. Alphab-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2010 Jul;69(7):694-703. doi: 10.1097/NEN.0b013e3181e4939c. PMID: 20535035. Veroni C, Serafini B, Rosicarelli B, Fagnani C, Aloisi F. Transcriptional profile and Epstein-Barr virus infection status of laser-cut immune infiltrates from the brain of patients with progressive multiple sclerosis. J Neuroinflammation. 2018 Jan 16;15(1):18. doi: 10.1186/s12974-017-1049-5. PMID: 29338732; PMCID: PMC5771146. Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 1985; 17: 278–82. doi: 10.1002/ana.410170310. Yeung MSY, Djelloul M, Steiner E, Bernard S, Salehpour M, Possnert G, Brundin L, Frisén J. Dynamics of oligodendrocyte generation in multiple sclerosis. Nature. 2019 Feb;566(7745):538-542. doi: 10.1038/s41586-018-0842-3. Epub 2019 Jan 23. Erratum in: Nature. 2019 Feb 5;: PMID: 30675058; PMCID: PMC6420067. Younger DS. Postmortem Neuropathology in Covid-19. Brain Pathol. 2020 Oct 23:e12915. doi: 10.1111/bpa.12915. Epub ahead of print. PMID: 33098141; PMCID: PMC7645938. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain. 2017 Jul 1;140(7):1900-1913. doi: 10.1093/brain/awx113. PMID: 28541408; PMCID: PMC6057548. Zrzavy T, Machado-Santos J, Christine S, Baumgartner C, Weiner HL, Butovsky O, Lassmann H. Dominant role of microglial and macrophage innate immune responses in human ischemic infarcts. Brain Pathol. 2018 Nov;28(6):791-805. doi: 10.1111/bpa.12583. Epub 2017 Dec 28. PMID: 29222823; PMCID: PMC6334527.
Copyright: © 2021 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |