|
|
|
Free Neuropathology 1:28 (2020) |
|
Original Paper |
|
Quantitative proteomic profiling of white matter in cases of cerebral amyloid angiopathy reveals upregulation of extracellular matrix proteins and clusterin |
|
Antigoni Manousopoulou1**, Ho Ming Yuen2**, Matthew MacGregor Sharp2, Satoshi Saito2, Roxana Aldea3, Norman Mazer3, Spiros D. Garbis4, Roxana O. Carare2* |
|
1 Beckman Research Institute, City of Hope, Duarte, CA, USA |
|
Corresponding author: |
|
Submitted: 14 August 2020 Accepted: 06 October 2020 Copyedited by: Biswa Ramani Published: 08 October 2020 |
|
Additional resources and electronic supplementary material: supplementary material (zip) |
|
Keywords: Extracellular matrix, Cell adhesion, Clusterin, Cerebral amyloid angiopathy, CAA, White matter, Proteomics |
|
Abstract
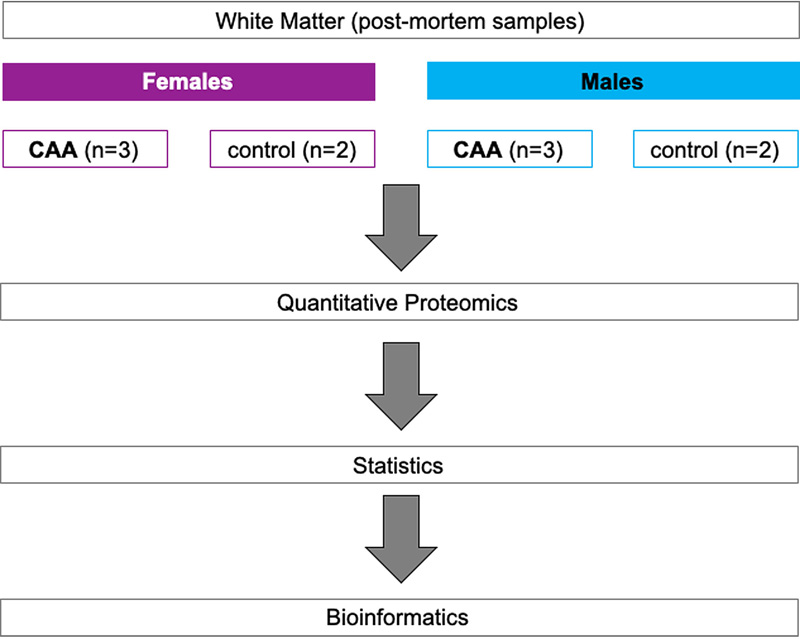
Aims: Cerebral amyloid angiopathy (CAA) is the accumulation of amyloid beta (Aβ) in the walls of cerebral arterioles, arteries and capillaries. Changes in the white matter in CAA are observed as hyperintensities and dilated perivascular spaces on MRI suggesting impairment of fluid drainage but the pathophysiology behind these changes is poorly understood. We tested the hypothesis that proteins associated with clearance of Aβ peptides are upregulated in the white matter in cases of CAA. Abbreviations CAA - cerebral amyloid angiopathy, WMH - white matter hyperintensities, IPA - Ingenuity pathway analysis, COL4A2 - Collagen IV alpha 2, HSPG2 - heparan sulfate proteoglycan 2, EMILIN2 - EMI Domain Endowed, TLN1 - Talin1, CLU - Clusterin, IDE - Insulin degrading enzyme, DSP - Desmoplakin, IPAD - Intramural Periarterial Drainage. Introduction Cerebral amyloid angiopathy (CAA) is characterized by the deposition of amyloid proteins including amyloid beta (Aβ), cystatin C, prion protein, ABri/ADan, transthyretin, gelsolin and immunoglobulin light chain amyloid (1) in the walls of leptomenigeal arteries and cortical arterioles (CAA-Type 2) (2) and in the walls of arteries or capillaries (CAA-Type 1), but rarely in the walls of venules (1-3). Sporadic CAA is a common feature of Alzheimer’s disease (1-3) and reflects the failure of clearance of proteins and interstitial fluid from the ageing brain (4, 5). Although CAA is uncommon in the white matter, (6) there are radiological features in the white matter in cases of CAA that include white matter hyperintensities (WMH) and dilated perivascular spaces (7, 8). The underlining pathophysiology is poorly understood, but the WMH are associated with small vessel disease (9). In addition to damage to nerve fibres particularly in the deep white matter, the WMH suggest that there is an increase in the volume of interstitial fluid (ISF) in the affected white matter. Pathologically, WMH are characterised by pallor of myelin staining in the affected white matter with morphological changes in astrocytes and axons and disruption of the distribution of aquaporin 4 in astrocyte processes (10). Imaging studies in the early stages of WMH suggest that the lesions are due to altered interstitial fluid mobility and water content that may be reversible (11). Interstitial fluid and Aβ enter the basement membranes of capillaries and arteries to be cleared from the brain along the intramural periarterial drainage (IPAD) pathways (12, 13). With increasing age, the structure of basement membranes changes and Aβ accumulates in the walls of arteries as CAA (14-16). Our proteomic studies performed on leptomeningeal arteries from brains with CAA demonstrated an upregulation of clusterin (apolipoprotein J) and tissue inhibitors of metalloproteinases 3 (TIMP3) (17). The aim of the present study was to compare the quantitative proteomic profile of white matter from patients with CAA to the corresponding brain region of age-matched controls in order to gain insight in the pathophysiology of white matter changes in CAA and to identify novel therapeutic targets. An overview of the present study workflow is presented in Figure 1.
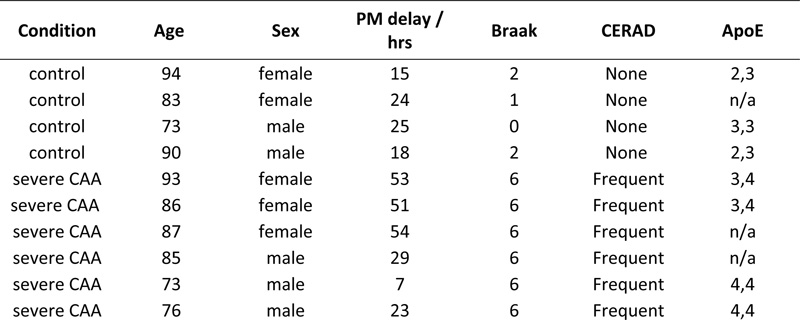
Figure 1: Outline of experimental workflow. Labelled peptides were analysed using two-dimensional liquid chromatography and tandem mass spectrometry. Materials and methods Tissue samples Fresh frozen white matter from the occipital lobe of severe CAA cases (n=6) and non-demented aged matched controls (n=4) was supplied by Newcastle Brain Tissue Resource (Ethics REC 08/H0906/136) (Table 1). All cases were diagnosed according to internationally-used criteria including neuritic Braak stages (18), Thal amyloid phases (19), and CERAD scores (20) (Table 1). Table 1: Header
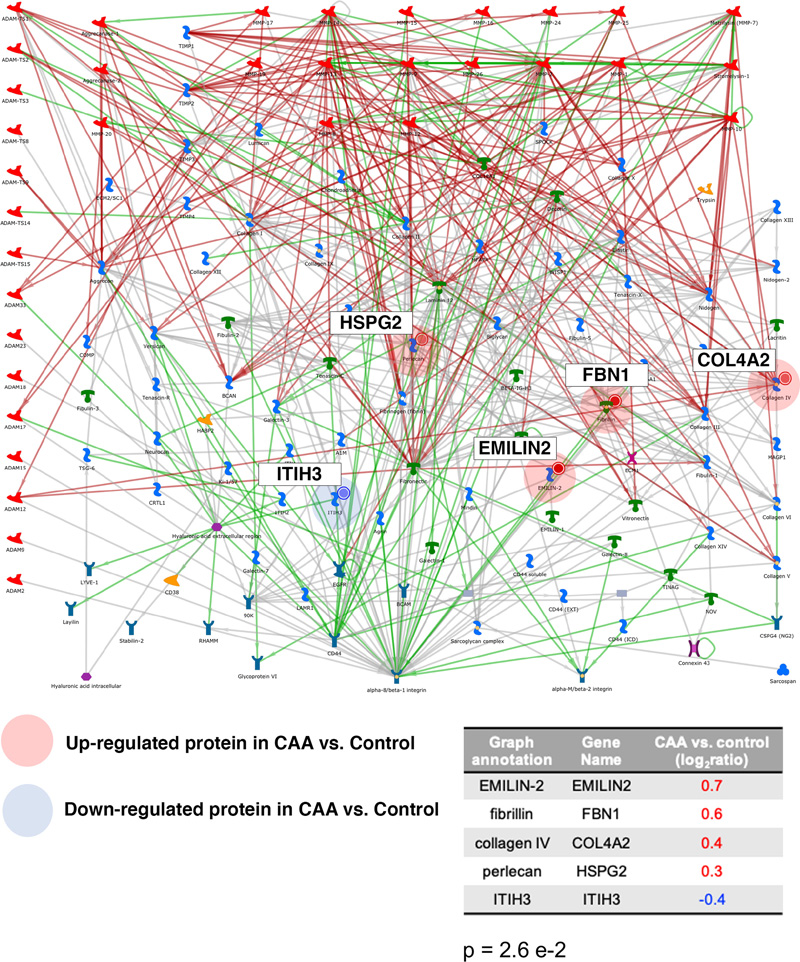
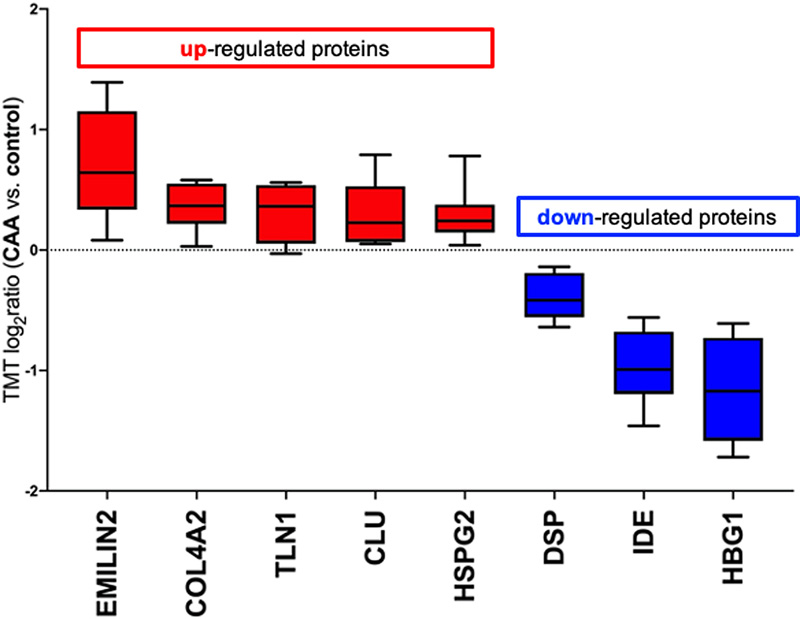
Table 1: Demographics of the cases used for this study. PM: post-mortem. The PM delay refers to the number of hours post-mortem until the tissue was dissected and frozen. Tissue quantitative proteomics analysis Samples of 1mg per case were dissolved in 0.5 M triethylammonium bicarbonate in 0.05% sodium dodecyl sulphate. These were then subjected to pulsed probe sonication (Misonix, Farmingdale, NY, USA) and lysates were centrifuged (16,000 g, 10 min, 4°C). Supernatants were measured for protein content using the bicinchoninic acid assay (Pierce BCA Protein Assay Kit, Thermo Fisher Scientific, Waltham, MA, US). 100 μg of protein was used per sample, adjusted to the highest volume. Proteins were reduced with 4μL 50mM Tris(2-carboxyethyl)phosphine hydrochloride at 60°C for 1 hour and alkylated using 2 μL 200mM methanethiosulfonate at room temperature for 10 min. Proteins were enzymatically digested using trypsin overnight in dark at 37°C (Sigma Aldrich, St. Louis, MO, USA). Peptides from each sample were labelled using the 10plex Tandem Mass Tag (TMT) reagent kit (Thermo Fisher Scientific, Waltham, MA, US). Labelled peptides were mixed and analyzed using two-dimensional liquid chromatography and tandem mass spectrometry as reported previously (17). Database searching Unprocessed raw files were submitted to Proteome Discoverer 1.4 (Thermo Fisher Scientific, Waltham, MA, US) for target decoy search using Sequest. The UniProtKB homo sapiens database which comprised 20,159 entries (release date January 2015) was utilized. The search allowed for up to two missed cleavages, a precursor mass tolerance of 10ppm, a minimum peptide length of six and a maximum of two variable (one equal) modifications of; oxidation (M), deamidation (N, Q), or phosphorylation (S, T, Y). Methylthio (C) and TMT 6-plex (K, Y and N-terminus) were set as fixed modifications. False discovery rate (FDR) corrected p value at the peptide level was set at <0.05. Percent co-isolation excluding peptides from quantitation was set at 50. Reporter ion ratios from unique peptides only were taken into consideration for the quantitation of the respective protein. Quantitation per protein in each of the six CAA patients was divided by each of the two matched same sex controls, which yielded 12 quantitative ratios per protein. These ratios, which represent the overall fold change in protein in the CAA patients with respect to the controls, were median-normalized and log2 transformed. We then combined the two respective log2 ratios per CAA patient when compared to the two controls by their mean which generated one observation per CAA patient per protein (see Supplementary Table 1). Due to small sample size, statistical analysis was performed per protein regardless of gender (n=6). Based on a previous study (16), we set the cut point of log2 ratio higher than or equals to 0.6, or lower than or equals to -0.6 to identify differentially expressed proteins (DEPs) (equivalent to a 1.5-fold change), but this was considered too restrictive for complex structure such as the white matter (number of proteins included = 51). Hence, we have narrowed the gap by halving the cut point to ±0.3 (equivalent to 1.2-fold change) instead in this study (number of proteins included = 189). Proteins identified with at least two unique peptides were included in the analysis. One-sample t test was performed on these log2 ratios per protein, comparing to 0, to identify the proteins that were statistically significant and differentially expressed in CAA patients comparing to controls (p <0.05). Bioinformatics analysis Ingenuity Pathway Analysis (IPA) (Qiagen, Hilden, Germany) and MetaCore (Clarivate Analytics, Philadelphia, PA, USA) were used to identify significantly enriched processes and pathways in the differentially expressed proteins of white matter from patients with CAA vs. control. P values in the context of canonical pathway analysis reflect how significantly the pathway is over-represented in the dataset of DEPs in white matter from patients with CAA vs. controls. Significance was set at p < 0.05. Results The global, untargeted proteomic analysis resulted in the profiling of 3,734 proteins (peptide FDR p<0.05) (Supplementary Tables 1, 2, 3). Of these, 189 were differentially expressed in CAA (Supplementary Table 4). Bioinformatics analysis of the differentially expressed proteins (DEPs) using MetaCore showed significant enrichment of the cell adhesion | cell-matrix interaction process (Figure 2). Mapping on this process network, EMILIN-2 (EMILIN2), fibrillin-1 (FBN1), collagen alpha-2(IV) (COL4A2) and the extracellular protein heparan sulfate proteoglycan (HSPG2) were upregulated; inter-alpha trypsin inhibitor heavy chain H3 (ITIH3) was downregulated in CAA vs. control. Figure 3 is a bar graph showing the relative expression levels of key upregulated (EMILIN2, COL4A2, talin-1 (TLN1), clusterin (CLU)) and downregulated (desmoplakin (DSP), insulin-degrading enzyme (IDE), hemoglobin subunit gamma-1 (HBG1)) proteins in the white matter of patients with CAA vs. controls (Figure 3).
Figure 2: Bioinformatics analysis using MetaCore showed significant enrichment of the cell-adhesion | cell-matrix interaction processes in the differentially expressed proteins of white matter from patients with CAA vs. controls. Upregulated proteins (EMILIN-2, Fibrillin, Collagen IV, Perlecan) are highlighted with a red circle whereas downregulated proteins (ITIH3) with a blue circle. The p represents significance of enrichment (i.e. this pathway is significantly enriched in the list of differentially expressed proteins).
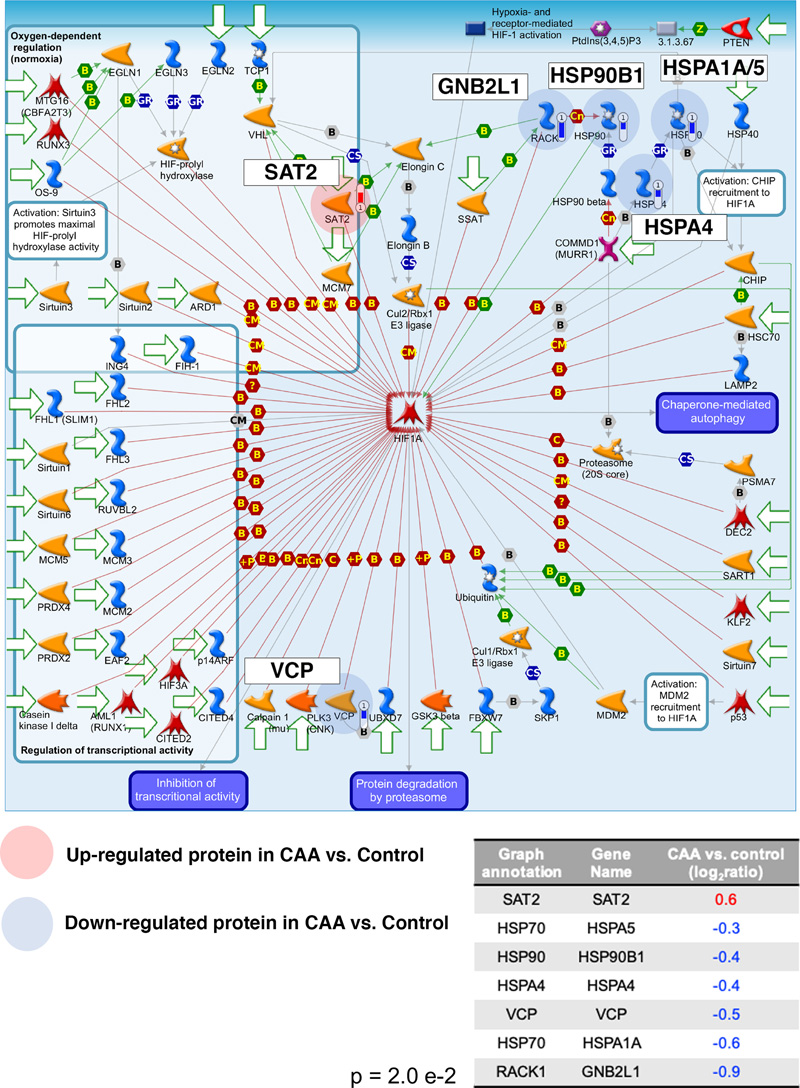
Figure 3: Up-regulated (red) and downregulated (blue) proteins in white matter from patients with CAA vs. controls. A positive log2ratio (ratio>1) reflects up- whereas a negative log2ratio (ratio<1) reflects downregulation of the respective protein in patients with CAA vs. controls. MetaCore demonstrated dysregulation of the hypoxia-inducible factor 1alpha (HIF1A) in CAA with upregulation of diamine acetyltransferase 2 (SAT2) and downregulation of heat shock 70kDa protein 4 (HSPA4), heat shock 70kDa protein 1A/1B (HSPA1A), 78kDa glucose-related protein (HSPA5), endoplasmin (HSP90B1), transitional endoplasmic reticulum ATPase (VCP) and guanine nucleotide binding protein subunit beta-2-like-1 (GNB2L1), suggesting altered response to hypoxia in CAA (Figure 4).
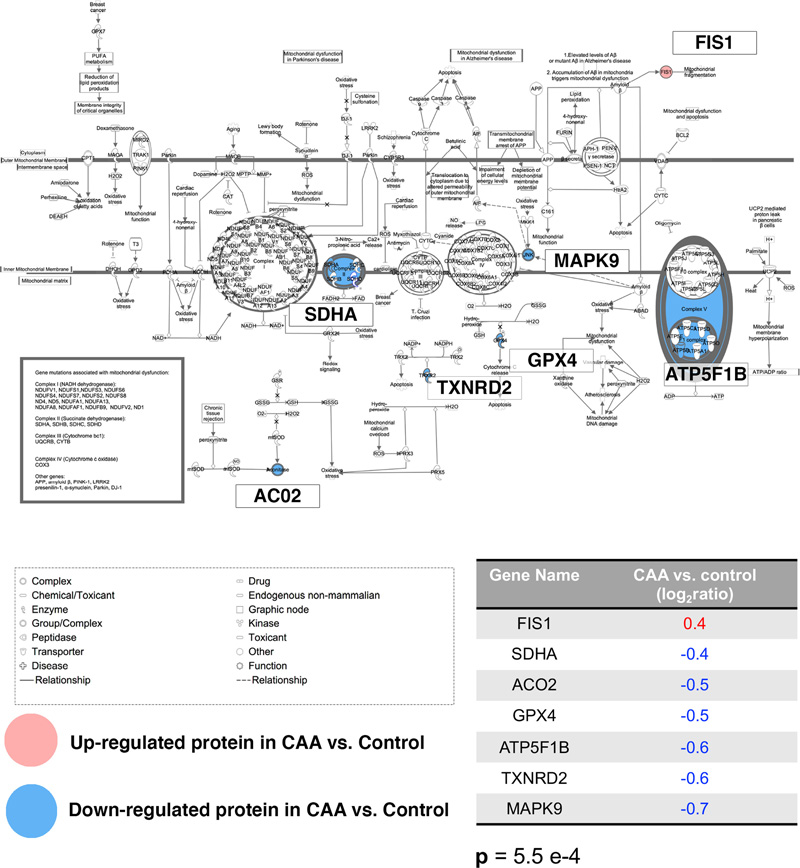
Figure 4: Negative regulation of HIF1A was a significantly enriched pathway map in the differentially expressed proteins of white matter from patients with CAA vs. controls. Upregulated proteins are highlighted with a red circle whereas downregulated proteins with a blue circle. More specifically, SAT2 was upregulated whereas HSP70, HSPA4, HSP90, RACK1 were downregulated in CAA vs. control. The p represents significance of enrichment (i.e. this pathway is significantly enriched in the list of differentially expressed proteins). Mitochondrial function also appeared altered in CAA with upregulation of the mitochondrial fission 1 protein (FIS1), a marker of mitochondrial and mitophagy (21) and reductions in proteins associated with the normal function of the mitochondria (mitochondrial succinate dehydrogenase flavoprotein subunit (SDHA), mitochondrial aconitate hydratase (ACO2), mitochondrial phospholipid hydroperoxide glutathione peroxidase (GPX4), mitochondrial ATP synthase subunit beta (ATP5F1B), mitochondrial thioredoxin reductase 2 (TXNRD2), mitogen-activated protein kinase 9 (MAPK9)). Mitochondrial dysfunction was a significantly over-represented canonical pathway (Figure 5).
Figure 5: Mitochondrial dysfunction was a significantly enriched canonical pathway in the differentially expressed proteins of white matter from patients with CAA vs. controls. Upregulated proteins are denoted with red whereas downregulated proteins with blue. More specifically, FIS1 was upregulated whereas SDHA, ACO2, GPX4, ATP5F1B, TXNRD2 and MAPK9 were downregulated in CAA vs. control. The p represents significance of enrichment (i.e. this pathway is significantly enriched in the list of differentially expressed proteins). Discussion The present study reports a comprehensive global proteomic profile of post-mortem white matter samples from patients with CAA compared to the corresponding brain region of age-matched controls. Out of 189 proteins identified as differentially expressed with both Metacore and IPA, several proteins associated with mitochondrial dysfunction, hypoxia, and clearance of Aβ are significantly up- or downregulated. The pathways reflecting hypoxia and mitochondrial dysfunction have been observed in previous analyses of the white matter in Alzheimer’s disease (22). Hypoxia-inducible factor-1 (HIF1) regulates oxygen homeostasis and thus has a major role in the control of energy/metabolism and angiogenesis. Diamine-acetyltransferase-2 (SAT2), guanine nucleotide binding protein subunit beta-2-like 1 (GNB2L1) and heat shock proteins are involved in the degradation of HIF1 (23, 24) and they were expressed at altered levels in the white matter of CAA cases (Figure 4). In the present study there was an increase in FIS1, a marker of mitochondrial and mitophagy (21), and a reduction in proteins associated with the normal function of the mitochondria (SDHA, ACO2, GPX4, ATP5F1B, TXNRD2, MAPK9), suggesting an overall dysregulation of the functions of the mitochondria in the white matter from cases of CAA (Figure 5). Most of the novel proteins showing substantial changes in the present study of the white matter in CAA were proteins associated with the extracellular matrix constituents and clearance of Aβ. Proteins with increased expression Collagen IV alpha 2 chain (COL4A2) is a highly conserved protein across species and is present in almost all vascular basement membranes (BM) (25). A common variation in the COL4A2 gene is associated with patients with intracerebral haemorrhage (26), which is one of the most frequent CAA-related cerebrovascular complications. Consistent with the current proteomic results, previous studies have reported increased collagen IV expression in cerebral microvessels in patients with Alzheimer’s disease (27) and increased collagen IV immunostaining in the leptomeningeal arteries in hereditary Cystatin C amyloid angiopathy (28). A recent proteomic study of the grey matter in AD with CAA type 1 (predominantly capillary CAA) has identified collagen alpha-2(VI) (COL6A2) as increased in the grey matter of CAA Type 1(29). Perlecan is a heparan sulfate proteoglycan 2 (HSPG2), also a component of BM and provides dynamic flexibility to the BM, while promoting the aggregation of Aβ (30, 31). Pathological studies have reported increased expression of HSPG2 in Alzheimer’s disease (32) with a significant association of HSPG2 polymorphism in apolipoprotein ε4 allele carriers (33). The upregulation of COL4A2 and HSPG2 have been observed in other studies in the grey matter, but their increase in the white matter of CAA cases in the present study demonstrate the widespread changes in the extracellular matrix in the brain, reflected in a failure of intramural periarterial drainage (IPAD). EMILIN-2 was expressed at higher levels in the white matter from brains with CAA vs. controls. EMILIN-2 is part of the EDEN (EMI Domain ENdowed) spectrum of proteins (34), with roles in angiogenesis, as well as cell adhesion/migration (34, 35). EMILIN-2 possesses the gC1q domain, which interacts with α4β1 integrin, which in turn binds to fibronectin, a major component of BM (36). Recent studies demonstrated a reduction in fibronectin in post-mortem brains with white matter hyperintensities as well as on cerebrovascular smooth muscle cell cultures exposed to hypercapnia as a model of hypoperfusion (37). Although the expression of EMILIN-2 in brain is relatively small compared to other organs and the physiological role of EMILIN-2 is unclear (38), its association with BM (39) suggests that the upregulation of EMILIN-2 reflects a compensatory upregulation of angiogenesis and IPAD in the face of the hypoperfusion and altered extracellular matrix characteristics of the white matter in CAA (15, 40). Talin-1 (TLN1) is a heavy protein that contains a C-terminal flexible rod domain which binds to vinculin and actin while the N-terminal head binds to integrin cytoplasmic tails (41-43). Binding of the talin head to integrin is a key step required for integrin activation (44, 45). Integrins are essential for cell adhesion and α5β1 is anti-apoptotic and a receptor for Aβ (46). As talin also promotes axon growth (47), it is possible that it is upregulated as a compensatory effect of axonal and synapse destruction in white matter lesions (22). In the present study, the expression of CLU (clusterin) was higher in the white matter of CAA cases compared to controls. Our own prior proteomic study of leptomeningeal arteries in CAA revealed a high expression of CLU, and a recent study on the proteomic expression of grey matter in CAA also revealed an increased expression of CLU (17). Genome-wide association studies identified variations in CLU expression associated with Alzheimer’s disease (48, 49). Clusterin may act as a chaperone molecule that facilitates intramural periarterial drainage of soluble Aβ (50). Increased expression of CLU may be a compensatory mechanism to the cerebrovascular accumulation of Aβ in these cases. Proteins with decreased expression IDE (Insulin-degrading enzyme) was significantly decreased in our study. IDE is a key enzyme that degrades Aβ in the extracellular spaces (51). In diabetes mellitus there is inhibition of Aβ degradation directly associated with a reduced activity of IDE (52-54). Overexpression of IDE ameliorates Aβ pathology (55). IDE is expressed within the vessel walls and its activity is decreased in CAA (56). The decreased expression of IDE may reflect a failure of the proteolysis and consequent increased toxicity of Aβ. The expression of HBG1, the γ(A)-globin subunit of fetal haemoglobin, was also decreased in our study. HBG1 is capable of strongly binding to Aβ (57). An animal model of CAA exhibited impaired Aβ transcytosis from brain to blood, resulting in decreased level of Aβ in the blood (58). Decreased expression of HBG1 may result from reduced level of the Aβ and HBG1 complex in the blood in cases of CAA. Desmoplakin (DSP) is a desmosome-associated transmembrane glycoprotein and maintains the integrity of the epidermis and myocardium (59). The physiological role of desmoplakin in the brain remains unknown. Interestingly, it was reported that desmoplakin is a specific marker of lymphatic vessels in human tongue (60). Since IPAD represents the lymphatic drainage pathways of the brain parenchyma, (61) a decreased level of desmoplakin may simply reflect impaired IPAD. The present study has some limitations: Firstly, the sample size is low due to the difficulty in obtaining fresh frozen white matter. This has also prevented us from performing an analysis by sex or any other stratification. Secondly, it was not possible to specifically select areas of WMH from the frozen samples of brain and to study them separately. Nevertheless, our study has revealed generalised changes in the white matter in cases of CAA. Thirdly, we have not performed validation of the results by immunocytochemistry for the proteins involved. However, we have provided evidence from other studies that support our findings and provide a working hypothesis. While previous proteomic studies in the white matter in Alzheimer’s disease have identified modifications in proteins that are involved in cell survival and cell adhesion (22), in our study, we highlight that in the white matter of severe cases of CAA there is upregulation of the proteins that mark cell-matrix interaction, hypoxia, mitochondrial dysfunction and intramural periarterial drainage of soluble Aβ. The study from Castano et al in 2013 used AD cases with a very low CAA score (2 out of a maximum of 12) and identified GFAP, tropomyosins, calmodulin, annexin 1 and alpha-internexin as significantly upregulated, while ubiquitin carboxyl-terminal esterase and fascin-1 were down-regulated (22). The upregulated proteins are part of the cytoskeleton maintenance, calcium metabolism and cell survival pathways. We also have identified annexin and ubiquitin carboxyl-terminal esterase modified in the same pattern. The different results between the two studies are likely to be due to the very different CAA status of the cases; the present study points to pathways that are involved in the clearance of Aβ along the vessel walls. Our results suggest a working hypothesis that there is upregulation of the intramural periarterial drainage (IPAD) pathways in the white matter as a compensatory mechanism to the failure of drainage of interstitial fluid and solutes from the white matter and the grey matter that is a key pathogenetic factor in CAA. Acknowledgments The authors thank the Newcastle Brain Tissue Research Facility, the patients and their families. Ethical Approval Newcastle Brain Tissue Resource (Ethics REC 08/H0906/136). Author Contributions Antigoni Manousopoulou performed the proteomic experiments, analysed data, interpreted data and wrote manuscript; Roxana O. Carare and Spiros D. Garbis designed the study; Ho Ming Yuen assisted with statistical analysis; Matthew MacGregor Sharp, Satoshi Saito, Norman Mazer and Roxana Aldea had important intellectual contributions to the design of the study and analysis. References 1. Jellinger KA, Attems J. Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease 1. J NeurolSci. 2005;229-230:37-41. 2. Banerjee G, Carare R, Cordonnier C, Greenberg SM, Schneider JA, Smith EE, et al. The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry. . 2017;88(11):982-94. 3. Schreiber S, Wilisch-Neumann A, Schreiber F, Assmann A, Scheumann V, Perosa V, et al. Invited Review: The spectrum of age-related small vessel diseases: potential overlap and interactions of amyloid and nonamyloid vasculopathies. Neuropathol Appl Neurobiol. 2020;46(3):219-239. 4. Carare RO, Hawkes CA, Jeffrey M, Kalaria RN, Weller RO. Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobio. 2013;39(6):593-611. 5. Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol. 2015;25(1):63-78. 6. Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118(1):115-30. 7. Maniega SM, Valdes Hernandez MC, Clayden JD, Royle NA, Murray C, Morris Z, et al. White matter hyperintensities and normal-appearing white matter integrity in the aging brain. Neurobiol Aging. 2015;36(2):909-18. 8. Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol. 2020;16(3):137-53. 9. Al-Mashhadi S, Simpson JE, Heath PR, Dickman M, Forster G, Matthews FE, et al. Oxidative Glial Cell Damage Associated with White Matter Lesions in the Aging Human Brain. Brain Pathol. 2015;25(5):565-74. 10. Chen A, Akinyemi RO, Hase Y, Firbank MJ, Ndung'u MN, Foster V, et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Brain. 2016;139(Pt 1):242-58. 11. Wardlaw JM, Valdes Hernandez MC, Munoz-Maniega S. What are White Matter Hyperintensities Made of? Relevance to Vascular Cognitive Impairment. J Am Heart Assoc. 2015;4(6). 12. Morris AW, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A, et al. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016;131(5):725-36. 13. Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018;136(1):139-152. 14. Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011;121(4):431-43. 15. Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, et al. Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochimica et biophysica acta. 2016;1862(5):1037-46. 16. Hawkes CA, Gatherer M, Sharp MM, Dorr A, Yuen HM, Kalaria R, et al. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-beta from the mouse brain. Aging cell. 2013;12(2):224-36. 17. Manousopoulou A, Gatherer M, Smith C, Nicoll JA, Woelk CH, Johnson M, et al. Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2017 Oct;43(6):492-504. 18. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239-59. 19. Thal DR, Rub U, Schultz C, Sassin I, Ghebremedhin E, Del Tredici K, et al. Sequence of Abeta-protein deposition in the human medial temporal lobe. Journal of neuropathology and experimental neurology. 2000;59(8):733-48. 20. Morris JC, Mohs RC, Rogers H, Fillenbaum G, Heyman A. Consortium to establish a registry for Alzheimer's disease (CERAD) clinical and neuropsychological assessment of Alzheimer's disease. Psychopharmacol Bull. 1988;24(4):641-52. 21. Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochimica et biophysica acta. 2008;1777(7-8):860-6. 22. Castano EM, Maarouf CL, Wu T, Leal MC, Whiteside CM, Lue LF, et al. Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochemistry international. 2013;62(2):145-56. 23. Baek JH, Liu YV, McDonald KR, Wesley JB, Hubbi ME, Byun H, et al. Spermidine/spermine-N1-acetyltransferase 2 is an essential component of the ubiquitin ligase complex that regulates hypoxia-inducible factor 1alpha. J Biol Chem. 2007;282(32):23572-80. 24. Liu YV, Semenza GL. RACK1 vs. HSP90: competition for HIF-1 alpha degradation vs. stabilization. Cell Cycle. 2007;6(6):656-9. 25. Khoshnoodi J, Pedchenko V, Hudson BG. Mammalian collagen IV. Microsc Res Tech. 2008;71(5):357-70. 26. Rannikmae K, Davies G, Thomson PA, Bevan S, Devan WJ, Falcone GJ, et al. Common variation in COL4A1/COL4A2 is associated with sporadic cerebral small vessel disease. Neurology. 2015;84(9):918-26. 27. Kalaria RN, Pax AB. Increased collagen content of cerebral microvessels in Alzheimer's disease. Brain research. 1995;705(1-2):349-52. 28. Snorradottir AO, Isaksson HJ, Kaeser SA, Skodras AA, Olafsson E, Palsdottir A, et al. Deposition of collagen IV and aggrecan in leptomeningeal arteries of hereditary brain haemorrhage with amyloidosis. Brain research. 2013;1535:106-14. 29. Hondius DC, Eigenhuis KN, Morrema THJ, van der Schors RC, van Nierop P, Bugiani M, et al. Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer's disease. Acta Neuropathol Commun. 2018;6(1):46. 30. Trout AL, Rutkai I, Biose IJ, Bix GJ. Review of Alterations in Perlecan-Associated Vascular Risk Factors in Dementia. Int J Mol Sci. 2020;21(2). 31. Castillo GM, Ngo C, Cummings J, Wight TN, Snow AD. Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer's disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J Neurochem. 1997;69(6):2452-65. 32. Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer's disease. Neuropathol Appl Neurobiol. 2017;43(2):167-82. 33. Iivonen S, Helisalmi S, Mannermaa A, Alafuzoff I, Lehtovirta M, Soininen H, et al. Heparan sulfate proteoglycan 2 polymorphism in Alzheimer's disease and correlation with neuropathology. Neurosci Lett. 2003;352(2):146-50. 34. Colombatti A, Spessotto P, Doliana R, Mongiat M, Bressan GM, Esposito G. The EMILIN/Multimerin family. Front Immunol. 2012;2:93. 35. Paulitti A, Andreuzzi E, Bizzotto D, Pellicani R, Tarticchio G, Marastoni S, et al. The ablation of the matricellular protein EMILIN2 causes defective vascularization due to impaired EGFR-dependent IL-8 production affecting tumor growth. Oncogene. 2018;37(25):3399-414. 36. Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A. The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J Cell Biol. 2002;159(6):1071-86. 37. MacGregor Sharp M, Saito S, Keable A, Gatherer M, Aldea R, Agarwal N, et al. Demonstrating a reduced capacity for removal of fluid from cerebral white matter and hypoxia in areas of white matter hyperintensity associated with age and dementia. Acta Neuropathol Commun. 2020;8(1):131. 38. Doliana R, Bot S, Mungiguerra G, Canton A, Cilli SP, Colombatti A. Isolation and characterization of EMILIN-2, a new component of the growing EMILINs family and a member of the EMI domain-containing superfamily. J Biol Chem. 2001;276(15):12003-11. 39. Amma LL, Goodyear R, Faris JS, Jones I, Ng L, Richardson G, et al. An emilin family extracellular matrix protein identified in the cochlear basilar membrane. Mol Cell Neurosci. 2003;23(3):460-72. 40. Okamoto Y, Yamamoto T, Kalaria RN, Senzaki H, Maki T, Hase Y, et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 2012;123(3):381-94. 41. Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27:321-45. 42. Ye F, Lagarrigue F, Ginsberg MH. SnapShot: talin and the modular nature of the integrin adhesome. Cell. 2014;156(6):1340- e1. 43. Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol. 2013;14(8):503-17.4. Goult BT, Xu XP, Gingras AR, Swift M, Patel B, Bate N, et al. Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: implications for talin activation. J Struct Biol. 2013;184(1):21-32. 45. Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302(5642):103-6. 46. Matter ML, Zhang Z, Nordstedt C, Ruoslahti E. The alpha5beta1 integrin mediates elimination of amyloid-beta peptide and protects against apoptosis. J Cell Bio. 1998;141(4):1019-30. 47. Tan CL, Kwok JC, Patani R, Ffrench-Constant C, Chandran S, Fawcett JW. Integrin activation promotes axon growth on inhibitory chondroitin sulfate proteoglycans by enhancing integrin signaling. J Neurosci. 2011;31(17):6289-95. 48. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41(10):1094-9. 49. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41(10):1088-93. 50. Wojtas AM, Kang SS, Olley BM, Gatherer M, Shinohara M, Lozano PA, et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc Natl Acad Sci U S A . 2017;114(33):E6962-E71. 51. Saito S, Ihara M. New therapeutic approaches for Alzheimer's disease and cerebral amyloid angiopathy. Front Aging Neurosci. 2014;6:290. 52. Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer's disease: review and hypothesis. Neurobiol Aging. 2006;27(2):190-8. 53. Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169-78. 54. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162-7. 55. Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40(6):1087-93. 56. Morelli L, Llovera RE, Mathov I, Lue LF, Frangione B, Ghiso J, et al. Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid {beta} vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J Biol Chem. 2004;279(53):56004-13. 57. Perry RT, Gearhart DA, Wiener HW, Harrell LE, Barton JC, Kutlar A, et al. Hemoglobin binding to A beta and HBG2 SNP association suggest a role in Alzheimer's disease. Neurobiol Aging. 2008;29(2):185-93. 58. Davis J, Xu F, Miao J, Previti ML, Romanov G, Ziegler K, et al. Deficient cerebral clearance of vasculotropic mutant Dutch/Iowa Double Aβ in human AβPP transgenic mice. Neurobiol Aging. 2006;27(7):946-54. 59. Kam CY, Dubash AD, Magistrati E, Polo S, Satchell KJF, Sheikh F, et al. Desmoplakin maintains gap junctions by inhibiting Ras/MAPK and lysosomal degradation of connexin-43. J Cell Biol. 2018;217(9):3219-35. 60. Ebata N, Nodasaka Y, Sawa Y, Yamaoka Y, Makino S, Totsuka Y, et al. Desmoplakin as a specific marker of lymphatic vessels. Microvasc Res. 2001;61(1):40-8. 61. Engelhardt B, Carare RO, Bechmann I, Flugel A, Laman JD, Weller RO. Vascular, glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol. 2016;132(3):317-38.
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |