|
|
|
Free Neuropathology 1:3 (2020) |
|
Review |
|
Top ten discoveries of the year: Neuroinflammation |
|
Hans Lassmann |
|
Center for Brain Research, Medical University of Vienna, Vienna, Austria |
|
Corresponding author: |
|
Submitted: 25 December 2019 Accepted: 08 January 2020 Published: 11 January 2020 |
|
Keywords: Neuroimmunology, Brain inflammation, Lymphocytes, Microglia, Autoantibodies, Multiple sclerosis |
|
Abstract Ten neuropathological studies, published in 2019, are discussed, which address important aspects of neuroimmunology and inflammatory brain disease. They include topics related to new mechanisms of inflammation and immune mediated neurodegeneration, which are relevant for multiple sclerosis (publications 1 to 4) and discuss the role of specific autoantibodies against myelin oligodendrocyte glycoprotein or aquaporin 4 in neuromyelitis optica spectrum disorders (publications 5 and 6). Other studies highlight the discovery of new virus induced diseases of the nervous system and their relevance for clinical neurology and diagnostic neuropathology (publications 7 and 8). Finally, very interesting studies are discussed dealing with microglia and immune mechanisms in neurodegeneration (publication 9) and the neuropathological long-term outcome of Aß vaccination in Alzheimer’s disease (publication 10). All these studies highlight the central role of neuropathology in neurological disease research. Introduction There is a common belief that it is modern technology in molecular neurobiology, immunology and genetics, which drives progress in the understanding, diagnosis and therapy of human diseases of the nervous system, while neuropathology is sometimes considered an old fashioned and descriptive discipline which has little to add. However, very important new insights for human disease, achieved during the last years, came from neuropathological studies. In the field of neuroim-munology this included the discovery of new disease entities, new insights into the mechanisms of immune surveillance of the brain, of brain inflammation and of inflammation induced tissue damage in the nervous system. Although all these discoveries were based on a combined multidisciplinary approach, the interpretation of the structural changes in the brain tissue in human disease and experimental models, based on neuropathological competence and experience, was essential to draw the correct conclusions. Here I will discuss some recent work published during the last months, which contributed to a shift in the understanding of disease mechanisms. 1. Neuronal vulnerability and multilineage diversity in multiple sclerosis (Schirmer et al 2019) This study is based on a potentially revolutionary technical development. Up to now gene expression was performed either on homogenized tissue, which did not discriminate spatial, temporal or cellular changes during lesion development, or by in situ hybridization, which generally only provided information on the expression of single genes. Recently new technologies of single nucleus RNA sequencing together with multi-color or multi-channel-based methods of in situ hybridization and immunohistochemistry have been developed, which promise to overcome some of the above listed limitations. The study by Schirmer et al (2019) is one of the first to apply such technology for the analysis of multiple sclerosis (MS) brain tissue and combined this investigation with detailed confirmation by in situ hybridization and immunohistochemistry. The study supports the importance of meningeal inflammation with high B-lymphocyte content for cortical lesions, the heterogeneous astrocyte and microglia activation in the lesions, the loss of oligodendrocytes, the phagocytosis of myelin debris in macrophages during lesion activity and the importance of oxidative injury as a mechanism of neuronal injury. The most innovative finding is that a distinct subpopulation of excitatory neurons is dominantly affected in subpial cortical lesions of MS patients. Thus, this study impressively shows that this new technology can be applied on autopsy material of the human brain, and that the results reliably confirm what has been described with conventional technology before (Mahad et al 2015, Reich et al 2018). Even the main finding of a selective loss of a subset of excitatory neurons in the cortex is not surprising, since these cells are dominantly located in the outer cortical layers, which have been described before to show the most severe neuronal loss in subpial lesions (Magliozzi et al 2010). A similar approach has been used recently to determine oligodendrocyte heterogeneity in normal human controls and multiple sclerosis patients (Jäkel et al 2019). This study, too, showed that this new technology is suitable for the analysis of post mortem tissue of human inflammatory brain diseases and it reveals a new dimension regarding the complexity of cell phenotypes already in the normal tissue, being even more complex in multiple sclerosis lesions. However, to draw firm conclusions related to disease pathogenesis such studies have to be performed on much larger samples of patients and lesions and on the basis of much more elaborate selection and characterization of different lesion types. Thus, in future studies such technologies have to be applied on cases and lesions, which have been very carefully selected according to the specific research question. Unfortunately, availability of such material is very limited when the technology requires the use of fresh frozen tissue. Thus, major efforts are necessary to adapt these new technologies to formaldehyde fixed and paraffin embedded archival material. 2. Epstein-Barr Virus specific CD8 T cells selectively infiltrate the brain in multiple sclerosis and interact locally with virus-infected cells: Clue for a virus driven immunopathological mechanism (Serafini et al 2019) Even in the absence of an overt inflammatory disease of the central nervous system there is a small to moderate number of T-lymphocytes present in the brain and spinal cord, while infiltration of the tissue by T-cells and B-cells is massively increased in inflammatory brain diseases. From experimental data in normal mice and mouse models of neurodegenerative diseases or autoimmune encephalomyelitis it was suggested that these cells are MHC Class II restricted CD4+ T-cells, which enter the brain in the course of immune surveillance and, when directed against autoantigens of the CNS, may exert both, disease-promoting as well as regulatory or protective functions. These concepts however, were difficult to harmonize with previous observations that the majority of T-cells in the normal human brain and in the CNS of patients with neurodegenerative disease are CD8+ MHC class I restricted T-cells, displaying the phenotype of tissue resident memory cells (Smolders et al 2018). Similarly, in the MS brain CD8+ T-cells with a phenotype of tissue resident memory cells prevail, where they show focally restricted activation in particular in active lesion (van Nierop et al 2017, Machado Santos et al 2018; Figure 1C). So far, such tissue resident memory cells have been found, defined and characterized in solid tissues, including the brain, and in models of virus infection, where they form a very effective barrier against re-infection by the same agent (Steinbach et al 2018). Thus, a key question is, what target antigen is recognized by the CD8+ tissue resident T-cells in the MS brain. Multiple sclerosis is regarded by immunologists as an autoimmune disease, driven by T-lymphocytes directed against myelin anti-gen(s). However, attempts to identify an MS-specific autoimmune response have so far failed (Hohlfeld et al 2016). Even when the investigation specifically focused on activated CD8+ T-cells in the MS lesions, no reactivity was found against the classical candidates of myelin directed autoimmunity, but a substantial number of isolated T-cells recognized epitopes from Epstein Barr virus (EBV; Van Nierop et al 2017, Serafini et al 2019). Furthermore, Serafini et al (2019) showed that these EBV reactive T-cells form close contacts with B-lymphocytes expressing epitopes of EBV related proteins, and these T-cells display CD 107 on their surface, suggesting the release of the content of their cytotoxic granules. Overall these data suggest that the chronic inflammatory response in the MS brains may in part be driven by a T-cell response against EBV (Serafini et al 2019). However, the question whether there is productive EBV expression in the MS brain is still controversial. It is described by some groups, while other groups using similar techniques in material from comparable disease phenotypes and stages failed to confirm the data (van Nierop et al 2017). The reason for these discrepancies is still unresolved (Lassmann et al 2011). Another recent study (Konjevic Sabolek et al 2019) describes the interaction of activated CD8+ cytotoxic cells with local macrophages, but not with B-cells, which are the potential target for EBV infection. Thus, the specific antigenic target of T-cells in MS lesions is still unresolved, but recent data indicate that the concept of autoimmun-ity against myelin or other CNS antigens may be wrong.
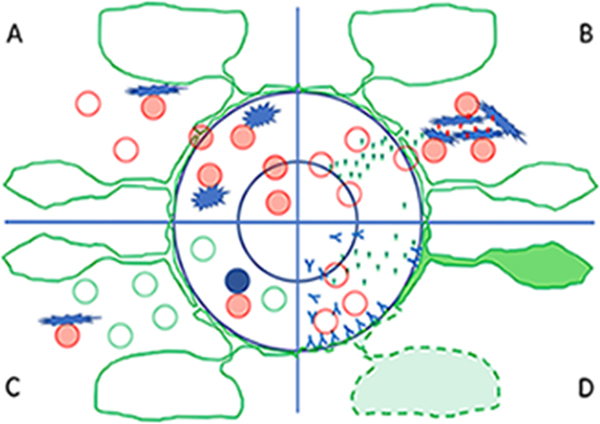
Figure 1: One well established and several new mechanisms triggering or propagating inflammation in the central nervous system (CNS) A: The basic well-established mechanism of immune surveillance and inflammation of the central nervous system is that T-cells, activat-ed in the peripheral immune system, are able to pass the blood brain barrier and enter the CNS in the process of immune surveillance. When they encounter their specific antigen, presented in the meninges, the perivascular space or the CNS parenchyma they get re-activated, produce proinflammatory cytokines and attract additional immune cells into the lesion. Red cells: activated T-cells, red ring cells: resting T-cells; star like cells: macrophages and microglia involved in antigen presentation; the blue circles indicate the endotheli-um of the blood brain barrier and the glia limitans. B: An alternative way to start inflammation in the CNS (as described by publication 3) is the local presence of damaged tissue or foreign antigens from infectious agents (red dots); they interact with microglia and induce inflammasome activation and the production of pro-inflammatory cytokines (green triangles). These cytokines activate endothelial cells at the blood brain barrier and allow the recruitment of T-cells into the focal site of tissue injury. In case these T-cells (red circles) find their cognate antigen within the lesion, they become activated (red cells) and trigger the full-blown inflammatory lesion. C: In conditions of chronic brain inflammation, such as for instance in multiple sclerosis (as discussed in context with publication 2), high numbers of tissue resident memory T-cells (green circles) are present within the brain and lesions and they are mainly in an inactivated stage. When there is re-appearance of their cognate antigen (for instance a foreign antigen or a modified self-antigen) presented by perivascular B-cells (blue circles) or by macrophages / microglia (blue stars), they get re-activated and propagate the chronic inflammatory process. D: A new mechanism of brain inflammation is outlined in publication 6. When high titers of antibodies against aquaporin 4 are present in the circulation, the very low amount of antibodies which can pass the normal blood-brain barrier, is sufficient to induce astrocyte dysfunction and their pro-inflammatory activation (green cells). This leads to the production of pro-inflammatory cytokines (green triangles), which activate the endothelium and massively disturb the blood brain barrier. This allows more antibodies together with leukocytes (red circles) and complement to enter the perivascular space, to destroy the astrocytes at the glia limitans and to promote inflammation. 3. Microglia nodules provide the environment for pathogenic T cells in human encephalitis (Tröscher et al 2019) A key question in the pathogenesis of inflammatory brain diseases is, why and how T-cells enter the brain and initiate the lesions. One mechanism is that they are activated in the peripheral immune system and get access to the brain in the course of immune surveillance (Wekerle et al 1986; Figure 1A). The other possibility, which may be particularly important in conditions of virus infection or neurodegeneration, is that local cells in the brain, such as for instance microglia, sense focal tissue injury and provide signals which drives the initial recruitment of CD8+ T-cells into the CNS (Figure 1B). Rasmussen’s encephalitis is a rare inflammatory seizure disorder mediated by cytotoxic T-cells which attack neurons and astrocytes (Bauer et al 2012). In the study by Tröscher et al (2019) it is shown that the initial stage of lesion formation is characterized by microglia activation and the formation of microglia nodules, which occurs before the first CD8+ T-cells have entered the brain. The activated microglia initially show inflammasome activation and up-regulation of endosomal Toll-like receptors, which is followed by the production of chemokines, the expression of major histocompatibility complex class I antigen, the activation of interferon signaling pathways and the recruitment of CD8+ T-lymphocytes. These data suggest that the initial trigger of inflammatory lesions in Rasmussen’s encephalitis is not the passage of activated T-cells through the blood brain barrier in the course of immune surveillance, but a local cell infection or tissue damage, which creates a focal pro-inflammatory environment through inflammasome activation. Subsequently, a subset of these recruited T-cells has to recognize their cognate antigen within the CNS to propagate the inflammatory reaction. Such a scenario may play a major role in the induction of inflammation in viral diseases of the nervous system, but similar microglia activation and the formation of microglia nodules is also seen around active lesions in the MS brain (Burm et al 2016). 4. Post mortem multiple sclerosis lesion pathology is influenced by single nu-cleotide polymorphism (Fransen et al 2020) Irrespective of the specific antigen, recognized by the inflammatory cells in MS lesions, the incidence and severity of brain inflammation appears to be influenced by the genetic background of the patients. This is now well established from genome wide association studies (GWAS), which define MS as a disease with a highly polygenic background (International Multiple Sclerosis Genetics Consortium 2019). More than 200 gene loci have been found associated with disease incidence, each of them with only very little individual impact. The global interpretation of these findings is that MS, as described before by pathology, is a chronic inflammatory disease of the nervous system, and its likelihood to affect a patient is influenced by multiple genes which in their interaction may enhance pro-inflammatory immune mechanisms. It is likely that such genetic polymorphisms also modify the pathological evolution of MS lesions, but considering their low impact on the disease in the general MS population, it was unexpected that such an effect may become apparent in autopsy cohorts. Fransen et al (2020) addressed this question on a material of 179 MS brains, containing a very high number of very well staged and characterized individual MS lesions. On this basis they were able to show that certain variants of immune related genes, which were before shown to be associated with disease severity, were also associated with the proportion of active lesions. The respective genes were in part related to apoptosis (FAS), T-cell activation (CTLA4) or are playing a role both in inflammation and neurodegeneration (CLEC16A). This study provides a first hint on how the genetic background of MS patients may affect pathological outcome of the lesions. However, there are still controversial issues regarding the statistical design for such investigations. Performing genotype/phenotype correlations with multiple potential risk associ-ated gene polymorphisms and multiple different pathological outcomes requires enormous stringency related to multiple testing, and there is some doubt that this can ever be reached with the limited autopsy material available. 5. Characterization of human myelin oligodendrocyte glycoprotein antibody response in demyelination (Tea et al 2019) Pathological studies performed during the last decade suggest that immunological mechanisms responsible for the induc-tion of MS-like inflammatory demyelinating lesions are heterogeneous between patients, in particular at the early stages of the disease and in patients with aggressive disease course (Lucchinetti et al 2000). One subtype of lesions was defined by immunoglobulin and complement deposition in active lesions with initial stages of demyelination. This was interpreted as evidence for the involvement of pathogenic autoantibodies, as it is reflected in models of experimental autoimmune encephalomyelitis, in which demyelination is triggered by antibodies against myelin oligodendrocyte glycoprotein (MOG) on the background of a T-cell mediated inflammatory reaction in the brain and spinal cord. New technologies, using trans-fected cell lines, are now available to identify potential pathogenic autoantibodies and using these techniques it was re-cently possible to identify a subset of patients with inflammatory demyelinating disease with a systemic antibody response against MOG (Borisow et al 2018). Identification of such demyelinating anti-MOG antibodies is difficult, since they are directed against complex conformational epitopes, while antibodies directed against conventional linear MOG epitopes are not pathogenic. The study by Tea et al (2019) describes the results of a detailed epitope mapping, which provides new information on the relevant target epitopes, recognized by potentially pathogenic MOG auto-antibodies. This is important for diagnosis of MOG antibody associated inflammatory demyelinating disease and for further analysis of pathogenetic mechanisms involved in antibody mediated demyelination. The identification of pathogenic MOG-antibodies is a major breakthrough in multiple sclerosis research, since current data suggest that the presence of anti-MOG antibodies defines an inflammatory demyelinating disease distinct from MS. It presents with clinical features of acute or relapsing disseminated encephalomyelitis or neuromyelitis optica and shows a different response to current anti-inflammatory treatments (Jarius et al 2016). These new findings further suggest that the experimental models of autoimmune encephalomyelitis more closely reflect MOG antibody associated disease than multiple sclerosis itself. 6. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat (Hillebrand et al 2019) Another disease, which originally has been classified as a variant of multiple sclerosis, is neuromyelitis optica (NMO). However, using similar techniques of auto-antibody detection as described above, it became clear that the majority of patients with NMO have auto-antibodies against the astrocytic water channel aquaporin 4 (AQP4). Further comprehensive clinical characterization of patients with AQP4 auto-antibodies enlarged the clinical phenotype, now designated as neuro-myelitis optica spectrum disorders (NMOSD), which are pathologically characterized by a primary inflammatory astro-cytopathy with secondary demyelination (Fujihara et al 2019). Experimental studies showed that patient derived auto-antibodies induce tissue damage in vitro or after direct injection into the brain, or when systemically injected into animals with T-cell mediated autoimmune encephalomyelitis. The study by Hillebrand et al (2019) shows for the first time that circulating AQP4 directed autoantibodies alone can induce NMOSD like disease in rats, provided that the injection resulted in a very high titer of circulating antibodies with very high specificity and affinity. Circulating antibodies reached the CNS tissue not only through circumventricular organs but also by the physiological leakage through cerebral veins (Figure 1D). Thus, a very low degree of AQP4 antibody leakage through the intact blood brain barrier may target perivascular astrocytes at the perivascular glia limitans and may augment further blood-brain barrier damage in the course of antibody mediated damage of the astrocytic process. This property is unique for AQP4 auto-antibodies and not seen with other antibodies such as those directed against MOG. The study further shows that brain damage by the leakage of the antibody in circumventricular organs is very limited and not associated with complement activation, possibly by the expression of complement inhibitory proteins. In contrast, when present in the perivenous space AQP4 antibodies induce profound blood-brain barrier injury, leakage of complement, recruitment of granulocytes and the activation of macrophages, and this gives rise to the full pathological spectrum of NMOSD lesions. This study, thus, provides some explanation for the heterogeneous spectrum of pathology in different brain regions of NMOSD patients. 7. The neuropathology of fatal encephalomyelitis in human Borna virus infection (Liesche et al 2019) Accurate diagnosis of non-purulent lymphocytic encephalitis in humans is a challenge. When no positive identification of a specific virus infection is possible, a diagnosis of encephalitis of autoimmune or probable infectious cause is assigned (Venkatesan and Murphy 2018, Seilhean 2019). The study by Liesche et al (2019) defines a new entity of lymphocytic scle-rosing panencephalomyelitis, which is caused by infection with Borna disease virus 1 (BoDV-1). Although the disease may follow generalized immunosuppression, in the majority of cases it developed spontaneously. The study provides a clear account of the pathological features of this new disease, consisting of mononuclear cell infiltrates, edema, microglia acti-vation and the presence of eosinophilic spherical inclusion bodies. Virus RNA was detected in neurons, ependymal cells, astrocytes and oligodendrocytes, but not in lymphocytes, macrophages or microglia. The distribution of the lesions in the brain and spinal cord was variable ranging from a dominant brain stem encephalitis, to prominent affection of the hippo-campus or a multifocal or diffuse pathology in other cases. In addition, infection may also involve the peripheral nervous system. This study adds Borna virus infection to the list of potential causative agents driving an acute or subacute inflam-matory disease of the central and peripheral nervous system, which has to be included in the spectrum of clinical differen-tial diagnoses. How frequent this condition is in the global population of patients with encephalitis has to be determined in the future. 8. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice (Figueiredo et al 2019) Another recently discovered disease of the nervous system is associated with Zika virus infection. Although originally be-lieved to disturb brain development in the course of intrauterine infection (Counotte et al 2019), it became clear in the last years that it may also infect the adult human nervous system and give rise to inflammatory diseases of the central and peripheral nervous system (da Silva et al 2017). The underlying mechanisms were up to now not defined. In this study the authors show that Zika virus can infect neurons and replicate not only in the human, but also in the mouse brain tissue in vitro. This allowed to study in more detail the consequences of Zika virus brain infection. In the mouse it mainly accumu-lates in memory associated brain regions, such as for instance the hippocampus, where it induces disturbances of long-term potentiation and impairs memory. These effects are associated and possibly mediated by microglia activation with the up-regulation of pro-inflammatory cytokines and the complement components C1q and C3. In line with this view it was also found that blockade of microglia activation or neutralization of TNF-α prevented synapse and memory dysfunction. These findings suggest that analysis of cognitive disturbances in Zika virus infected patients should be performed in a more systematic manner. However, it has also to be noted that similar mechanisms of cytokine and microglia activation associated synapse and memory disturbance occur in many different inflammatory conditions of the central nervous system, including even experimental autoimmune encephalomyelitis (Musella et al 2016). It is, thus, rather a general consequence of brain exposure to pro-inflammatory cytokines. Whether the mechanisms inducing synaptic dysfunction are different and specific in Zika virus infected brain has to be clarified in the future by direct comparison.
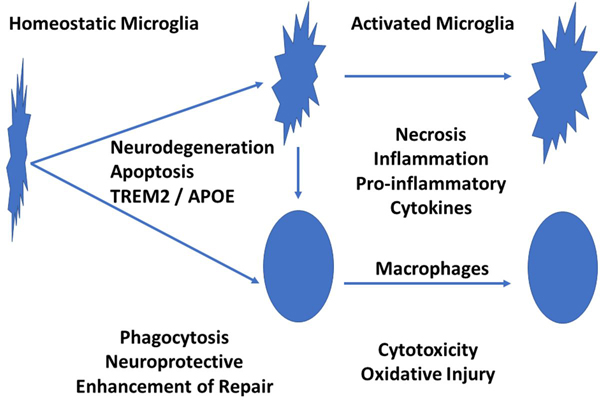
Figure 2: Microglia Activation in Neurodegeneration and Brain Inflammation The normal brain is populated by so called resting or homeostatic microglia. When they sense neurodegeneration, they become activat-ed through the TREM2/APOE pathway. In this activation state there are phagocytic cells responsible for the removal of tissue debris, which is essential to limit further injury and to allow tissue repair. When these activated microglia are further exposed to inflammatory cytokines, they differentiate into immunological effector cells, which are actively involved in tissue damage. Thus, both, an impairment of the initial step of microglia activation as well as the proinflammatory activation are detrimental for the brain. 9. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples (Prokop et al 2019) For many years our views on the pathogenesis of Alzheimer’s disease focused on protein misfolding and deposition of Aß and tau in the extracellular space or in the neuronal cytoplasm. In recent years, however, evidence accumulated that ele-ments of the innate immune system may play a role in promoting the cognitive disturbance in patients and the neuropa-thological damage in the brain. In particular, recent genome wide association studies identified polymorphisms of genes involved in microglia activation to be associated with the disease (Sims et al 2017). One of these genes is the TREM2 re-ceptor (Johnsson et al 2013), which when activated by neurodegeneration triggers initial microglia activation through the TREM2 / APOE pathway (Krasemann et al 2017). Although the role of these molecules in microglia activation in general and in experimental models of neurodegeneration is well understood, it is important to determine the neuropathological effects of the respective gene variants in Alzheimer’s disease patients. This question was addressed by the study of Prokop et al (2019). The authors describe that carriers of the respective TREM2 risk variants had lower numbers of plaque-associated microglia and this was associated with a higher degree of axonal injury and tau pathology in comparison to carefully matched Alzheimer’s disease patients without these TREM2 variants. In addition, microglia senescence was found to be enhanced and some global decrease in microglia activation in TREM2 variant AD cases was apparent. The study clearly underlines the difficulty of such an investigation in human autopsy material, since such differences only became apparent when exactly matched lesion stages were compared in patients with similar disease severity. This is due to the fact that these gene polymorphisms do not enable or prevent AD pathology, but just modify its speed of development. Overall, the data suggest that impairment of microglia activation not only enhances the progression of cognitive impairment, but also promotes axonal degeneration and tau pathology. Whether this is due to a reduced neuroprotective function of microglia or a reduced clearance of misfolded proteins due to impaired phagocytosis remains to be determined in the future. 10. Persistent neuropathological effects 14 years following amyloid-ß immunization in Alzheimer’s disease (Nicoll et al 2019) Following promising results, documenting the removal of amyloid plaques in mice after active immunization with Aß, a first clinical trial had been initiated almost 20 years ago, which was followed by several other trials using immunization strategies or the systemic application of specific Aß-antibodies (Wisniewski and Goni 2014, 2015). The first trial was stopped due to the appearance of autoimmune inflammatory disease of the central nervous system as a complication of the vaccination strategy. A detailed analysis of the neuropathology of patients who died in the years after the trial suggested that this vaccination resulted in clearance of Aß-deposits from the brain in association with the development of an Aß antibody response, but that this had no significant effect of the progression of dementia (Holmes et al 2008). The present study by Nicoll et al (2019) in detail describes the neuropathology in relation to the clinical outcome seen in 22 patients of this trial, who died within 14 years after the immunization. This comprehensive analysis provides important insights related to the design and outcome of Aß directed immunotherapies in Alzheimer’s disease. Only 77% of the patients included in this cohort had Alzheimer’s disease, while dementia in the others was due to different diseases. In the AD patients with Aß vaccination the vast majority provided evidence for plaque removal and the extent for amyloid deposition correlated inversely with the titer of antibodies directed against the N-terminal region of Aß, documenting the sustained effect of Aß vaccination reducing amyloid load. Despite a significantly lower extent of neurofibrillary pathology in patients with low amyloid load, there was still profound neurofibrillary tangle pathology in all cases. In addition, severe cerebral amyloid angiopathy was noted in the majority of patients, irrespective of the effect on Aß plaque removal. Progression of dementia was seen in most patients, even in those with extensive plaque removal. In conclusion, this study impressively documents the value of systematic neuropathological studies to monitor clinical dementia trials. It shows that Alzheimer’s disease patients can remain plaque-free even for 14 years after active immunization, but that this has little effect on tau pathology and the further development of dementia. Conclusion In this review a number of recent neuropathological studies are presented, which contributed to the dynamic change in our understanding of the pathogenesis of neuroinflammatory diseases. They highlight and confirm that the field of neuro-pathology occupies a central position in human disease research. Experimental models provide important insights into basic and molecular mechanisms, but their relevance for the disease in humans has to be determined by studying the disease in patients. Correct interpretation of the changes occurring in damaged tissues requires profound neuropathological expertise. References Bauer J, Vezzani A, Bien CG. Epileptoic encephalitis: the role of the innate and adaptive immune system. Brain Pathology 2012; 22:412-21 Borisow N, Mori M. Kuwabara S, Scheel M, Paul F. Diagnosis and treatment of NMO spectrum disorder and MOG encephalomyelitis. Front Neurol 2018; 9:888 Burm SM, Peferoen LA, Zuiderwijk-Sick EA, et al. Expression of Il-1ß in rhesus EAE and MS lesions is mainly induced in the CNS itself. J Neuroinflammation 2016; 13:138 Counotte MJ, Meili KW, Taghavi K, Calvet G, Low N. Zika virus infection as a cause of coingenital abnormalities and Guillain Barre syndrome. A living systematic review. F1000Res 2019; 8:1433 Da Silva IRF, Frontera JA, Bispo de Filippis AM, Nascimento OJMD. Neurologic complications associated with the Zika virus in Brazilian adults. JAMA Neurol 2017; 74:1190-1198 Figueiredo CP, Barris-Aragao FGQ, Neris RLS, Frost PS. Et al. Zika virus replicates in adult human tissue and impairs synapses and memory in mice. Nature Commun 2019; 10:3890 Fransen NL, Crusius JBA, Smolders J, Mizee MR et al. Post-mortem multiple sclerosis lesion pathology is influenced by single nucleotide polymorphisms. Brain Pathol 2020; 30:106-119 Fujihara K. Neuromyelitis optica spectrum disorders: still evolving and broadening. Curr Opin Neurol 2019; 32: 385-394 Hillebrand S, Schanda K, Nigritinou M, Tsymala I et al. Circulating AQP4-specific auto-antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol 2019; 137:467-485 Hohlfeld R, Dornmair K, Meinl E, Wekerle H. The search for the target antigens in multiple sclerosis, part 2: CD8+ T-cells, B-cells and antibodies in the focus of reverse ´translational research. Lancet Neurol 2016; 15: 317-31 Holmes C, Boche D, Wilkinson D, Yadegarfar G, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomized, placebo-controlled phase I trial. Lancet 2008; 372: 216-23 International Multiple Sclerosis Genetics Consortium. A systems biology approach uncovers cell-specific gene regulatory effects of genetic associations in multiple sclerosis. Nat Commun 2019; 10:2236 Jäkel S, Agirre E, Mendana Falcao A, van Bruggen D, et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019; 566: 543-547 Jarius S, Ruprecht K, Kleiter I, Borisow N et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment response and long-term outcome. J Neuroinflammat 2016; 13:280 Johnsson T, Stefanson H, Steinberg S, Jonsdottir I, et al. Variant of TREM2 associated with risk of Alzheimer’s disease. New Engl J Med 2013; 368: 107-116 Konjevic Sabolek M, Held K, Beltran E, et al. Communication of CD8(+) T cells with mononuclear phagocytes in multiple sclerosis. Ann Clin Transl Neurol 2019; 6:1151-1164 Krasemann S, Madore C, Cialic R, Baufeld C, et al. The TREM-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2018; 47:566-581 Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM; the NeuroproMiSe EBV Working Group. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue--report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria Brain 2011; 134: 2772-2786 Liesche F, Ruf V, Zoubaa S, Kaletka G et al. The neuropathology of fatal encephalomyelitis in human Borna virus infection. Acta Neuropathol 2019; 138: 653-665 Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination Ann Neurol 2000; 47: 707-717 Machado-Santos J, Saji E, Tröscher AR, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018; 141:2066-2082 Magliozzi R, Howell OW, Reeves C, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 2010; 68:477-493 Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015; 14:183-193 Musella A, Mandolesi G, Mori F, Gentile A, Centonze D. Linking synaptopathy and gray matter damage in multiple sclerosis. Mult Scler 2016; 22: 149-149 Nicoll JAR, Buckland GR, Harrison CH, Page A, et al. Persistent neuropathological effects 14 years following amyloid ß-immunization in Alzheimer’s disease. Brain 2019; 142: 2113-2126 Prokop S, Miller KR, Labra SR, Pitkin RM, et al. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropathol 2019; 138: 613-630 Reich DA, Lucchinetti CF, Calabresi PS. Multiple Sclerosis. New Engl J Med 2018; 378:169-180. Schirmer L, Velmeshev D, Holmqvist S, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019; 573:75-82 Seilhean D. Infections of the central nervous system: neuropathology. Rev Neurol (Paris) 2019; 175:431-435 Serafini B, Rosicarelli B, Veroni C, Mazzola GA, Aloisi F. Epstein-Barr virus-specific CD8 T cells selectively infiltrate the multiple sclerosis brain and interact locally with virus infected cells: clue for a virus-driven immunopathological mechanism. J Virol 2019; 93(24). pii: e00980-19. doi: 10.1128/JVI.00980-19. Sims R, van der Lee SJ, Naj AC, Bellenguez C, et al. Rare coding variants in PLC2, ABI3 and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 2017, 49: 1373-1384 Smolders J, Heutinck KM, Fransen NL, et al. Tissue-resident memory T cells populate the human brain. Nat Commun 2018; 9:4593 Steinbach K, Vincenti I, Merkler D. Resident-memory T cells in tissue restricted immune responses: For better or worse. Front Immunol 2018; 9:2827 Tea F, Lopez JA, Ramanathan S, Merheb V. et al. Characterization of human myelin oligodendrocyte glycoprotein antibody response in demyelination. Acta Neuropathol Commun 2019; 7:145 Tröscher A, Wimmer I, Quemada-Garrido L, Köck U, et al. (2019) Microglial nodules provide the environment for pathogenic T cells in human encephalitis Acta Neuropathol 2019; 137:619-635 van Nierop GP, van Luijn MM, Michels SS, et al. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol 2017; 134:383-401 Venkatesan A, Murphy OC. Viral encephalitis. Neurol Clin 2018; 36:705-724 Wisniewski T, Goni F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron 2015; 85: 1162-1176 Wekerle H, Linington C, Lassmann H, Meyermann R (1986) Cellular immune reactivity within the CNS. Trends Neurosci 1986; 9:271-277 Wisniewski T, Goni F. Immunotherapy of Alzheimer’s disease. Biochem Pharmacol 2014; 88: 499-507
Copyright: © 2020 The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited, a link to the Creative Commons license is provided, and any changes are indicated. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. |